fall_exp_extra4
advertisement

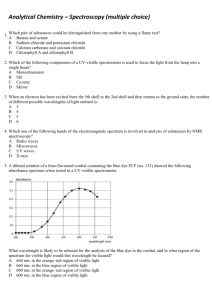
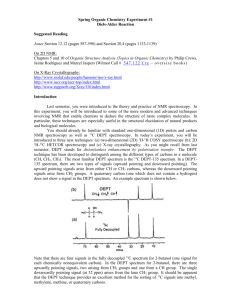
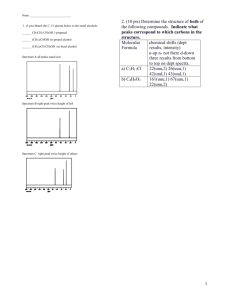
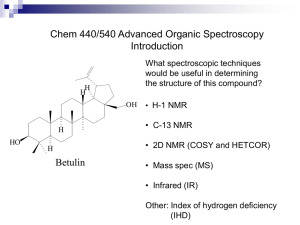
Fall Organic Chemistry Experiment #9 Advanced NMR Spectroscopy: Identification of Unknown Biological Compounds Introduction You were introduced to the theory and practice of NMR spectroscopy in experiment #6. In this experiment, you will be introduced to some of the more modern and advanced techniques involving NMR that enable chemists to deduce the structure of more complex molecules. In particular, these techniques are especially useful in the structural elucidation of biological molecules. You should already be familiar with standard one-dimensional (1D) proton and carbon NMR spectroscopy. In today’s experiment, you will be introduced to three new techniques: (a) 13 C DEPT spectroscopy; (b) two-dimensional (2D) COSY spectroscopy; and (c) 2D HETCOR spectroscopy. DEPT stands for distortionless enhancement by polarization transfer. The DEPT technique has been developed to distinguish among the different types of carbons in a molecule (CH, CH2, CH3). In a DEPT spectrum there are two types of signals (upward pointing and downward pointing). The upward pointing signals arise from either CH or CH3 carbons, whereas the downward pointing signals arise from CH2 groups. A saturated carbon (one which does not contain a hydrogen) does not show a signal in the DEPT spectrum. An example spectrum is shown below. Note that there are four signals in the fully decoupled 13C spectrum for 2-butanol (one signal for each chemically nonequivalent carbon). In the DEPT spectrum for 2-butanol, there are three upwardly pointing signals, two arising from CH3 groups and one from a CH group. The single downwardly pointing signal (at 32 ppm) arises from the lone CH2 group. It should be apparent that the DEPT technique provides an excellent method for the sorting of 13C signals into methyl, methylene, methine, or quaternary carbons. Pulsed NMR spectroscopy has enabled the chemist to devise experimental methods for the solution of complex organic structures. Traditional one-dimensional techniques do not allow the observer to discern complicate patterns when peak regions overlap. Such a situation normally defies elucidation by the interpreter. However, two-dimensional NMR techniques have arisen to allow for more definitive peak assignments and structural elucidation. A two-dimensional spectrum has two chemical shift axes and a third axis corresponding to peak intensity (a 1D plot has one shift axis and one intensity axis). The resultant spectrum when viewed from the side appears as a “mountain range”. Normally, however, we view the spectrum from the top in a form called a contour plot (see below). These contour plots (when correctly analyzed) show the correlation between neighboring nuclei in the molecule. The most common method involves a 1 H-1H shift correlation; that is, protons that are coupled to one another. This technique is called “correlation spectroscopy” or COSY. An example of a COSY spectrum is shown below. Remember, we view the plot from above (top view). Along the x-axis is a one-dimensional proton NMR of ethyl vinyl ether. Along the y-axis is another one-dimensional proton NMR of ethyl vinyl ether. All of the dots (peaks) that arise inside the box (contour plot) are significant peaks that will yield important structural information. Notice that some peaks lie along a diagonal while others are off of the diagonal. The peaks along the diagonal simply represent the individual peaks one each 1D proton NMR. The important peaks are the ones that are not along the diagonal. These peaks are the cross peaks. The cross peaks arise from pairs of protons that are splitting one another (that is, they are coupled or correlated). How do we use this information? The first thing we need to do is find the peaks along the diagonal and draw a line through them. Then, we can start at any cross peak (let’s use A) and draw a straight line vertically (up) to the diagonal and horizontally (over) to the diagonal. Notice that we have intersected the diagonal at two different peaks (1.1 ppm and 3.8 ppm). The protons giving rise to the peak at 1.1 ppm are from the terminal methyl group (labeled “a”). The protons giving rise to the peak at 3.8 ppm are from the methylene group (labeled “b”). Therefore, we can conclude that the cross peak A results from the correlation (coupling) of the methyl group protons with the neighboring methylene protons (as expected). The other cross peaks can be analyzed in a similar fashion. A second 2D NMR technique that also involves nuclear correlation is HETCOR or heteronuclear correlation spectroscopy. The appearance of a HETCOR spectrum is very similar to the COSY spectrum. Each axis along the perimeter represents a 1D spectrum. However, in HETCOR, one axis contains a 1D proton NMR and the other axis contains a 1D carbon NMR. The resultant HETCOR spectrum, therefore, indicates the coupling between protons and the carbon to which they are attached. For example, a HETCOR spectrum of 2-methyl-3-pentanone is shown below. Notice that a diagonal set of peaks does not exist in the example spectrum. A straight line is drawn from each cross peak to the x and y-axes. Notice in our example that cross peak A arises from the correlation of the carbon peak at 5 ppm on the x-axis and the proton peak at 0.9 ppm on the y-axis. We would conclude that the terminal methyl protons (of type “a”) are located on the carbon at the 5 ppm chemical shift. Certainly, each of these examples has not demonstrated the power of two-dimensional techniques for solving structures. However, you would be hard pressed to solve the structure of complex synthetic and biological molecules without employing one or both of these techniques. In fact, you will likely find that these techniques will be very helpful in elucidating the structure of your unknown in today’s experiment. Procedure You are to obtain a series of spectroscopic data (1H-NMR, 13C-NMR, 13C-DEPT, and 1H1 H -COSY, IR, and GCMS) for an unknown bicyclic[3.1.1]terpene. Prepare your sample by dissolving 75 mg of the unknown in ~1 mL of CDCl3/TMS. You’ll be responsible for the FULL interpretation of the data being sure to designate all of the coupling constants and type of coupling (construct a TABLE). You will also be asked to identify your unknown terpene. In addition, you will model each of the four possible terpenes (a-pinene, myrtenol, myrtenal, and verbenone) using PCSpartan Plus. You primary objective is to observe the relationship between dihedral angle versus coupling constants – known as the Karplus relationship (see WOC Experiment #19.10 on page 263). The Karplus relationship predicts that hydrogens with dihedral angles near 90° should approach a minumum value while those near 0° or 180° should be maximized. For more information on the Karplus relationship see: (1) Organic Structure Analysis by Philip Crews, Jaime Rodriguez, and Marcel Jaspers (2) Basic One and Two Dimensional NMR Spectroscopy by Horst Friebolin (3) Organic Structural Spectroscopy by Joseph Lambert, Herbert Shurvell, David Lightner, and R. Graham Cooks In addition, you’ll have to be on the look out for other types of coupling (e.g. vicinal, geminal, 4bond, five-bond, and W-coupling). Again, the references above should serve as excellent resources for these various categories of coupling. Be advised that these subtle coupling relationships often are the critical features that allow us to elucidate the structure of complex biological molecules! In order to effectively model these compounds, you will build them using PCSpartan Plus, minimize using the AM1 level and geometry optimization. Once minimized, you can determine all of the relevant dihedral angles (see table below). You’ll want to use the same numbering scheme as shown on the template structure below. NOTE: “s” means syn and “a” means anti – with respect to the gem-dimethyl bridge – you may find it helpful to build a structure using your hand-held model kit. Compound Verbenone Myrtenol Myrtenal a-Pinene H1-H7a H1-H7s H3-H4s H3H4a H4s-H5 H4a-H5 H5-H7a H5-H7s