Night Starvation Template - PBL-J-2015
advertisement

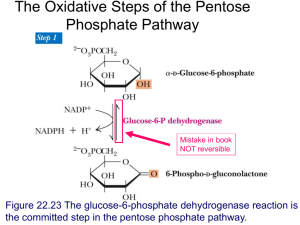
Week 4 - Night Starvation John - 3 y/o male: GLYCOGEN STORAGE DISEASE TYPE 1A Relevant Symptoms (incl. relevant negatives) PRESENTATION: 8PM Diaphoretic Tremulous Distressed Other Significant History Has vomited Last meal: 4pm (?did vomiting occur before or after last meal??) Hx of night sweats (independent of external temperature) Hx of previous admissions w/ hyperlipidaemia, lactic acidosis (>10mmol-1/L) and hypoglycaemia w/ improvement w/ IV dextrose 5%. Previous episodes associated with intercurrent febrile (infective) illness with anorexia and vomiting. Birth history normal; 50% centile for height and weight Examination and Signs Below 3rd centile for height and weight Diaphoretic Anxious Pallor Distended Abdomen Hepatomegaly palpable to umbilicus BGL: undetectable Improvement upon administration of IV glucose Test results: 1) Bloods: - Pin-prick BGL test: glucose levels undetectable - Previous blood results: hypoglycamia, lactic acidosis, hyperlipidaemia 2) Other: - Liver biopsy: Glucose-6-Phosphotase activity <5% of normal Provisional diagnosis: Inborn error of CHO metabolism: glycogen storage disease (type 1) Other possible tests: - Controlled fast with continuous blood readings - Liver ultrasound (rule out carcinoma) Differential Diagnosis: Other pathologies of glycogen storage Fructose 1,6 bisphosphotase deficiency Discussed: endocrine disorders (type one diabetes, hypothyroidism, hyperpituitarism), endocrine tumours (secreting pituitary adenoma), hepatocellular carcinoma, acute renal failure, adrenal insufficiency Clinical Reasoning: John presents to the emergency department acutely distressed. He is pale, sweaty, anxious and tremulous, indicating some sort of insult to the body provoking a sympathetic nervous response. With his normal birth history, and deteriorating growth rate, it is clear that John is failing to thrive. This must raise concerns of an ongoing problem. The history is very important. John has presented many times to the emergency department before with similar symptoms. The fact that he has often been febrile on acute presentations hints at acute exacerbations of his underlying condition due to incurrent illness. It is not clear whether John is currently ill due to some infectious cause, however his mother tells us that his last meal was at 4pm, four hours ago. John may or may not have vomited after this event, something we may wish to clarify with his mother. John's history of abnormal blood results (hyperlipidaemia, lactic acidosis and hypoglycaemia), combined with massive hepatomegaly is strongly suggestive of a metabolic illness. A liver biopsy clinches the diagnosis, revealing markedly decreased glucose-6-phosphatase activity. Aetiology In the post-prandial state (post-meal), insulin is secreted by beta pancreatic islet cells in order to promote the uptake of glucose and other nutrients from the blood into the tissues. In the fasted state, blood glucose homeostasis is maintained by the catabolism of stored liver (and renal) glycogen into glucose, via glucose-6phosphate, which is then released into the blood. Patients with glycogen storage disease type 1a cannot effectively dephosphorylate glucose-6-phosphate to release glucose into the blood, and thus present with hypoglycaemia, massive hepatomegaly and renomegaly due to an excess of stored glycogen. Histologically, hepatocytes are swollen with a clear cytoplasm as a result of the large amounts of glycogen in the cell. Because glucose-6-phosphate cannot be dephosphorylated, blood glucose cannot be effectively maintained between meals. Patients often present with hypoglycaemia and it's more severe sequellae, including neuroglycopaenia, seizures and coma. Repetitive episodes of hypoglycaemia result in the up-regulation of synthesis and transport of counter-regulatory (stress) hormones such as glucagon, cortisol, catecholamines and growth hormone, with predictable metabolic effects. Muscle glycolysis is often upregulated, resulting in high levels of pyruvate, lactate and free fatty acid being released into systemic circulation. In the liver, FFAs undergo beta oxidation to be converted into acetyl coA, which can then be used in the krebs cycle, as a medium for ketone body production, or in triglyceride resynthesis. Aditionally, fatty acids circulate in the blood as FFAs or triacylglycerol which may manifest itself with high levels of serum TGs, and later, lipid plaque deposits in lean tissue such as skeletal muscle, the heart, liver and pancreatic islet cells, resulting in lipotoxicity, with deleterious tissue effects (steato-hepatitis, insulin resistance, cardiac contractile dysfunction, and pancreatic beta cell failure/insulin deficiency). Hyperuricaemia results from the shifting of glucose-6-phosphate into the ribose-5-phosphate pathway of nucelotide biosynthesis, of which urea is a metabolic byproduct. Excessive urate is produced during the production of ATP from adenosin 5'-diphosphate in a reaction that involves deamination of AMP to inosine, with resultant conversion to uric acid. This hyperuricaemia is worsened by the excess lactate that competes with the uric acid for excretion by a common renal anion transporter. Thus, the clinical picture is one of: - Hepatorenomegaly (storage site of glycogen) - Metabolic acidosis as a result of a decreasing anion gap due to: (note these are all acidic products) o Lactic acid excess o o o Hyperuricaemia (uric ACID) Ketone body excess (KETOACIDS) FFA excess (FATTY ACIDS) SIMPLIFIED DIAGRAM OF HEPATIC FUNCTIONS: Risk Factors: - Family history of GSD 1a Affects 1 in 100,000 births More common in Ashkenazi Jews (1 in 68 = carrier) Management Plan Problem Hypoglycaemic Events Goal/desired outcome Euglycaemia Medications Mode of action Method (incl. patient actions) Frequent, low GI meals (3-4hrs apart) Diet: 65-70% CHO, <20% fat (equal amounts of PUFAs, MUFAs and SFAs), 10-15% protein; low cholesterol, reduce milk and fruit intake. Supply of low GI food nasogastrically overnight Frequent BGLs and Lactic acid levels Periodic (3-6m) assessment of growth, development and metabolic parameters (electrolytes, lactic acid, lipids, uric acid, aminotransferases, renal function, vitamin D, thyroid function and FBCs) Side effects Nil - consider lipid lowering drugs with advancing age (fibrate) Other Psychosocial/ethical/legal/patient-centred considerations LONG TERM SEQUELLAE TO GSD1a: - Hepatic adenomas develop by age 30 o May rupture, causing blood loss o May develop into hepatocellular carcinoma - Nephropathy Renal failure - Osteoporosis (due to effects of counter-regulatory hormones on bone density) - Bleeding tendency - Impairment of growth - Menstrual abnormalities and hirsuitism - Hypothyroidism - Vascular dysfunction Female patients have been reported to have conceived and delivered healthy pregnancies (MD Consult) PPH/PPD implications Medical Consultation (week 4) Chronic disease (Week 26) Resources/health professional s involved Patient + Family Dietician GP Endocrinologist Any specific monitoring required? Resources used/discovered