File - Medical Nutrition Therapy Manual
advertisement

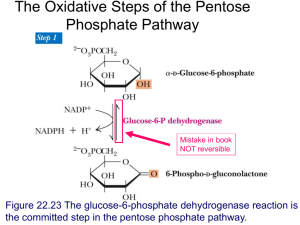
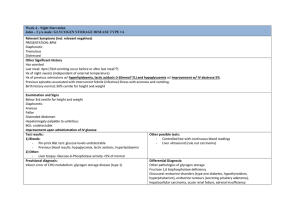
Claire Millonig KNH 413 23 April 2015 Glycogen Storage Disease Overview: There are 11 different types within the diagnosis of glycogen storage disease. However, the most common is type 1 glycogen storage, also known as Gierke’s disease. This is an inherited disorder that results in metabolism abnormalities. Normally, after we eat, the liver stores glycogen to keep our blood glucose levels normal but in the case of someone with type 1 glycogen storage disease, the enzyme needed to release glycogen from the liver is missing. Therefore, blood glucose levels drop into a hypoglycemic state within a few hours after eating. Glycogen storage disease is an inherited disorder, which results in a lack of the enzyme required to break down glycogen into glucose to be released into the body. Glycogen stores are used when a person is not eating to keep blood glucose levels stable. Since this is a genetic disorder, it is most commonly diagnosed in children between the ages of 4 and 10, sometimes even in infants (ALF). Symptoms include extreme hunger, delayed growth, fatigue, irritability, enlarged liver and a swollen abdomen. If these symptoms are observed, a child will most likely see his or her doctor for further testing. These tests include blood tests, ultrasounds to measure the liver and kidneys, and genetic testing or liver biopsy is possible. The blood tests will include an evaluation of blood glucose levels, cholesterol, triglycerides, lactate and uric acid. If a needle biopsy or liver biopsy were required, the child would need to be admitted to the hospital for these procedures. The main focus of treatment for type 1 glycogen storage disease is making sure the child continues to grow properly and correcting the metabolic changes in the body. Most commonly, raw cornstarch (1-2 g/kg body weight per dose) mixed with water, soy formula or soymilk is to be consumed every 3-6 hours (Nelms). The cornstarch digests slowly, which allows a slow and steady release of glucose. Also, small, frequent feedings are recommended throughout the day. The child may also require an enteral feeding tube at night to supply glucose steadily or a cornstarch mixture may be consumed before bed and once in the middle of the night. Most doctors agree that fructose and galactose should be limited in the diet, however the amount of restriction is done on a case-by-case basis. Since there are many limitations to what a child with type 1 glycogen storage disease can eat, it is recommended to take a multivitamin and possibly a calcium supplement. It is recommended that a patient with type 1 glycogen storage disease consume 60%-70% of their total calories from carbohydrates, 10%-15% from protein and less than 30% from fat (Nelms). In type 1 glycogen storage disease, infections are common because of a weakened immune system. If the child develops any kind of infection or sickness, an IV with glucose would be necessary until the issue is resolved. Because of the weakened immune system, adequate sources of zinc are necessary to help support a healthy and strong immune system as well as to support normal growth. Currently there is research being done about gene therapy in patients with type 1 glycogen storage disease, however, no conclusions have been drawn yet. Glycogen storage disease is diagnoses during infancy and childhood, which resulted in very little survivors through adulthood in the past. With more advanced medicine and diet therapy than before this is no longer the case. Careful monitoring of diet and blood sugar levels now allows people to live into adulthood. Without careful monitoring, low blood sugar can become fatal. It is important to be aware of infections, kidney complications, high blood pressure and tumors in the liver. Long-term complications are most commonly renal failure, kidney stones, osteoporosis (brittle bones), benign cysts on the ovaries, and benign tumors on the liver (American Liver Foundation). General Guidelines: Dietary treatment o Small, frequent meals throughout the day. o Consume 60%-70% of their total calories from carbohydrates, 10%-15% from protein and less than 30% from fat o Restricted intake of fructose and galactose Fruits, juice, milk, dairy products and sweets that contain sucrose o Multivitamin and calcium supplementation For young children o Raw cornstarch (1-2 g/kg body weight per dose) mixed with water, soy formula or soy milk every 3-6 hours. o Raw cornstarch mixture before bed and once in the middle of the night if not using enteral drip. Sickness o Enteral tube should be used for infusion of carbohydrates o If this is not tolerated, then they should be hospitalized to receive glucose through an IV. Additional Resources: American Liver Foundation: http://www.liverfoundation.org/abouttheliver/info/gsdi/ University of Florida: https://ufhealth.org/sites/default/files/media/GSD/GeneralNutrition-Guidelines-for-Glycogen-Storage-Disease-Type-I.pdf Sample Diet for a 2-year-old child: Time 8 am Food Sliced banana Amount ½ banana 10 am 12:30 pm 3 pm 6 pm 7:30 pm Cheerios Soy Milk Granola Bar Shredded deli turkey Cheese pieces Cooked carrot pieces Water Toast Almond Milk Green beans Rice Shredded Chicken breast Soy Milk Soy Milk Popcorn ½ cup 8 fl. Oz. 1 bar 1.5 oz. 1 slice ¼ cup 8 fl. Oz. 1 slice 8 fl. Oz. ½ cup ½ cup 2 oz. 4 fl. Oz. 4 fl. Oz. 1 cup Sources Genetics of Glycogen-Storage Disease Type I Treatment & Management. (n.d.). Retrieved March 9, 2015, from http://emedicine.medscape.com/article/949937-treatment Glycogen storage disease type Ib | Counsyl. (n.d.). Retrieved March 9, 2015, from https://www.counsyl.com/services/family-prep-screen/diseases/glycogenstorage-disease-type-ib/ Nelms, M. (2011). Nutrition therapy and pathophysiology (2nd ed.). Belmont, CA: Wadsworth, Cengage Learning. Type I Glycogen Storage Disease. (n.d.). Retrieved March 9, 2015, from http://www.agsdus.org/html/typeigsd.html Type I Glycogen Storage Disease. (n.d.). Retrieved March 9, 2015, from http://www.liverfoundation.org/abouttheliver/info/gsdi/ Zinc. (n.d.). Retrieved March 9, 2015, from http://ods.od.nih.gov/factsheets/ZincHealthProfessional/