Part II
advertisement

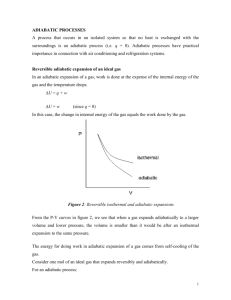
Chapter 5 Continued: More Topics in Classical Thermodynamics Einstein on Thermodynamics (1910) • “A theory is the more impressive the greater the simplicity of its premises, and the more extended its area of applicability.” Einstein on Thermodynamics (1910) • “A theory is the more impressive the greater the simplicity of its premises, and the more extended its area of applicability.” • “Classical Thermodynamics… is the ONLY physical theory of universal content which I am convinced that, within the applicability of its basic concepts, WILL NEVER BE overthrown.” Eddington on Thermodynamics (1929) • “If someone points out to you that your pet theory of the universe is in disagreement with Maxwell’s Equations – then so much the worse for Maxwell’s equations. • “But if your theory is found to be against the 2nd Law of Thermodynamics - I can offer you no hope; there is nothing for it but to collapse in deepest humiliation!” Free Expansion ( The Joule Effect) • A type of Adiabatic Process is the FREE EXPANSION in which a gas is allowed to expand in volume adiabatically without doing any work. • It is adiabatic, so by definition, no heat flows in or out (Q = 0). Also no work is done because the gas does not move any other object, so W = 0. The 1st Law is: Q = ΔE + W • So, since Q = W = 0, the 1st Law says that ΔE = 0. • Thus this is a very peculiar type of expansion and In a Free Expansion, The Internal Energy of a Gas Does Not Change! Free Expansion Experiment • Experimentally, an Adiabatic Free Expansion of a gas into a vacuum cools a real (non-ideal) gas. • The temperature is unchanged for an Ideal Gas. • Since Q = W = 0, the 1st Law says that ΔE = 0. • For an Ideal Gas it is easily shown that E is independent of volume V, so that E = E(T) = CTn (C = constant, n > 0) Free Expansion • For an Ideal Gas E = E(T) = CTn (C = constant, n > 0) • So, for Adiabatic Free Expansion of an Ideal Gas since ΔE = 0, ΔT = 0!! • Doing an adiabatic free expansion experiment on a gas gives a means of determining experimentally how close (or not) the gas is to being ideal. • T = 0 in the free expansion of an ideal gas. But, for the free expansion of Real Gases, T depends on V. • So, to analyze the free expansion of real gases, its convenient to Define The Joule Coefficient αJ (∂T/∂V)E (= 0 for an ideal gas) • Some useful manipulation: αJ (∂T/∂V)E = – (∂T/∂E)V(∂E/∂V)T – (∂E/∂V)T/CV Combined 1st & 2nd Laws: dE = T dS – pdV. • Joule Coefficient: αJ = – (∂E/∂V)T/CV • 1st & 2nd Laws: dE = T dS – pdV. • So (∂E/∂V)T = T(∂S/∂V)T – p. • A Maxwell Relation is (∂S/∂V)T = (∂p/∂T)V, so that the Joule coefficient can be written: αJ = (∂T/∂V)E = – [T(∂P/∂T)T – p]/CV Obtained from the gas Equation of State αJ is a measure of how close to “ideal” a real gas is! Joule-Thompson or “Throttling” Effect (Also Known as the Joule-Kelvin Effect! Why?) • An experiment by Joule & Thompson showed that the enthalpy H of a real gas is not only a function of the temperature T, but it is also a function of the pressure p. See figure. Adiabatic Wall Thermometers p1,V1,T1 p2,V2,T2 Porous Plug Throttling Expansion p1 > p2 • The Joule-Thompson Effect is a continuous adiabatic process in which the wall temperatures remain constant after equilibrium is reached. • For a given mass of gas, the work done is: W = p2V2 – p1V1. 1st Law: ΔE = E2 - E1 = Q – W. Adiabatic Process: Q = 0 So, E2 – E1 = – (p2V2 – p1V1). This gives E2 + p2V2 = E1 + p1V1. • Recall the definition of Enthalpy: H pV . • So in the Joule-Thompson process, the Enthalpy H stays constant: H2 = H1 or ΔH = 0. • To analyze the Joule-Thompson Effect its convenient to Define: • The Joule-Thompson Coefficient μ (∂T/∂p)H (μ > 0 for cooling. μ < 0 for heating) Some useful manipulation: μ = (∂T/∂p)H = – (∂T/∂H)P (∂H/∂p)T = – (∂H/∂p)T/CP. 1st & 2nd Laws: dH = TdS + V dp. 12 Joule-Thompson Coefficient μ (∂T/∂p)H (μ > 0 for cooling. μ < 0 for heating) Some useful manipulation: μ = (∂T/∂p)H = – (∂T/∂H)P (∂H/∂p)T = – (∂H/∂p)T/CP. The 1st Law: dH = TdS + Vdp. So, (∂H/∂p)T = T(∂S/∂p)T + V. A Maxwell Relation is (∂S/∂p)T = – (∂V/∂T)p so that the Joule-Thompson Coefficient can be written: μ = (∂T/∂p)H = [T(∂V/∂T)T – V]/CP Obtained from the gas Equation of State More on the Joule-Thompson Coefficient • The temperature behavior of a substance during a throttling (H = constant) process is described by the Joule-Thompson Coefficient, defined as • The Joule-Thompson Coefficient is clearly a measure of the change in temperature of a substance with pressure during a constant-enthalpy process, & we have shown that it can also be expressed as Throttling A Constant Enthalpy Process H = E + PV = Constant • Characterized by the Joule-Thomson Coefficient, which can be expressed as Another Kind of Throttling Process! (From American slang!) Throttling Processes: Typical T vs. p Curves Family of Curves of Constant H (Reif’s Fig. 5.10.3) 0 0 • Now, for a brief, hopefully useful Discussion of a Microscopic Physics Model in this Macroscopic Thermodynamics chapter! • Let the system of interest be a real (non-ideal) gas. An early empirical model developed for such a gas is The Van der Waals’ Equation of State • This is a relatively simple Empirical Model which attempts to make corrections to the Ideal Gas Law. Recall the Ideal Gas Law: pV = nRT The Van der Waals Equation of State has the form: (P + a/v2)(v – b) = RT v molar volume = (V/n), n # of moles • This model reproduces the behavior of real gases more accurately than the ideal gas equation through the empirical parameters a & b, which represent the following phenomena: • The term a/v2 represents the attractive intermolecular forces, which reduce the pressure at the walls compared to that within the gas. • The term – b represents the molecular volume occupied by a kilomole of gas, & which is therefore unavailable to other molecules. • As a & b become smaller, or as T becomes larger, the 19 equation approaches ideal gas equation Pv = RT. Adiabatic Processes in an Ideal Gas • • • • Ratio of Specific Heats: γ cP/cV = CP/CV. For a reversible quasi-static process, dE = dQ – PdV. For an adiabatic process, dQ = 0, so that dE = – P dV. For an ideal gas, E = E(T), so that CV = (dE/dT). Also, PV = nRT and H = E + PV, so that H =H(T). So, H = H(T) and CP = (dH/dT). • Thus, CP – CV = (dH/dT) – (dE/dT) = d(PV)/dT = nR. So, CP – CV = nR. • This is sometimes known as Mayer’s Equation, & it holds for ideal gases only. • For 1 kmole, cP – cV = R, where cP & cV are specific heats. 20 • Since dQ = 0 for an adiabatic process: dE = – P dV & dE = CV dT, so dT = – (P/CV) dV . • For an ideal gas, PV = nRT so that P dV +V dP = nR dT = – (nRP/CV) dV. & V dP + P (1 +nR/CV) dV = 0. This gives, CV dP/P + (CV + nR) dV/V = 0. For an ideal gas ONLY, CP – CV = nR. so that CV dP/P + CP dV/V = 0, or dP/P + γ dV/V = 0. • Simple Kinetic Theory for a monatomic ideal gas (Ch. 6) gets E = (3/2)nRT so Cv = (3/2)nR & γ = (Cp/Cv) = (5/3) • Integration of the last equation in green gives: ln P + γ ln V = constant, so that γ PV = constant. 21 Work Done on an Ideal Gas in a Reversible Adiabatic Process Method 1: Direct Integration • For a reversible adiabatic process, PVγ = K. • Since the process is reversible, W = PdV, so that W = K V–γ dV = – [K/(γ –1)] V–(γ–1) = – [1/(γ –1)] PV | (limits: P1V1 P2V2) • So, W = – [1/(γ –1)] [P2V2 – P1V1]. • For an ideal monatomic gas, γ = 5/3, so that W = –(3/2)] [P2V2 – P1V1]. 22 Work Done on an Ideal Gas in a Reversible Adiabatic Process Method 3: From the 1st Law • For a reversible process, W = Qr – ΔE. • So, for a reversible adiabatic process: W = – ΔE. • For an ideal gas, ΔE = CV ΔT = ncV ΔT = ncV (T2 – T1). So, for a reversible adiabatic process in an ideal gas: W = – ncV (T2 – T1). • For an ideal gas PV = nRT, so that W = – (cV/R)[P2V2 – P1V1]. • But, Mayer’s relationship for an ideal gas gives: R = cP – cV so that W = – [cV/(cP – cV)][P2V2 – P1V1] or W = – [1/(γ –1)] [P2V2 – P1V1]. 23 Summary: Reversible Processes for an Ideal Gas Adiabatic Process PVγ = K γ = CP/CV W= Isothermal Process Isobaric Process Isochoric Process T = constant P = constant V = constant W= – [1/(γ –1)] [P2V2 – P1V1] nRT ln(V2 /V1) ΔE = CV ΔT ΔE = 0 W = P V W=0 ΔE = CV ΔT ΔE = CV ΔT PV = nRT, E = ncVT, cP – cV = R, γ = cP/cV Monatomic ideal gas cV = (3/2)R, γ = 5/3 24