Loss-of-function mutations in the human homeobox gene
advertisement

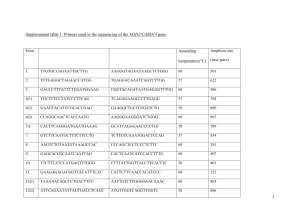
Supplementary Information Supplementary Information Methods Patients. Ethical approval was obtained from the Central Oxford Research Ethics Committee and skull phenotypes were assessed by plain radiographs, 3D computed tomography (CT) scanning or magnetic resonance imaging (MRI). No individuals had radiological manifestations of craniosynostosis or cleidocranial dysplasia, and at least one affected person from each family had a normal G-banded karyotype. Clinical examination of families segregating ALX4 mutations (pedigrees in Fig. 1b) did not demonstrate dysmorphic features or abnormalities of the limbs, abdominal wall, male genitalia, skin, hair or teeth. Family 1 is British. The proband (IV-1) presented as a neonate with a midline skull defect (width 8.5 cm at 7 months) and a patent vitellointestinal duct. Bilateral PFM <1 cm width were identified on skull radiography in his father III-3 (aged 40 y) and great uncle II-1 (aged 69 y), neither of whom were previously aware of their condition. Individual III-1, the daughter of II-1, was born with a midline skull defect and had unexplained drop attacks during childhood. A CT brain scan showed an enlarged cerebrospinal fluid (CSF)-filled space in the posterior fossa. Family 2 is of Slovakian origin; affection status, compared with the original pedigree7, was revised in some individuals after skull radiography. ALX4 heterozygotes had classical PFM, except for an obligate carrier aged 43 y (II-4) with increased bone thickness only, a boy aged 5 y (IV-5) with abnormal skull modelling and his sister aged 4 months (IV-6) who exhibited mild cranium bifidum. Two individuals had learning difficulties of unknown cause. Family 3 resides in Spain: a large posterior fontanelle 1 Supplementary Information was identified in III-6 by prenatal ultrasound and a midline skull defect was confirmed after birth8. Family 4 resides in Brazil. Apart from benign neonatal convulsions in II-2 and III-2, no additional clinical abnormalities were noted. Individual III-2 manifests cranium bifidum, as shown in Fig. 2a. CT and MRI brain scanning of individuals I-2, II2 and III-2 demonstrated a wide hiatus of the tentorium cerebelli associated with a large CSF-filled space, but no cerebellar hypoplasia. Measurements of the size of PFM (expressed as a percentage of the maximum skull width on PA or AP skull radiograph) were collated for all cases of known age with an identified mutation in MSX2 or ALX4 (refs 1,2,7,8,16–18). Exclusion of MSX2 mutations and analysis of linkage to 11p. We excluded mutations or large deletions of MSX2 in families 1–4 by linkage analysis, detection of intragenic heterozygosity and sequencing of the coding region as previously described1. Linkage of chromosome 11p simple sequence repeat polymorphisms including D11S1393, D11S903, D11S2095 and D11S554 (refs 6,19 and The Genome Database, http://www.gdb.org) to PFM was examined in families 2 and 3. The phenotype was considered as autosomal dominant with a mutant allele frequency of 10-5. We defined three liability classes with different penetrance values in heterozygotes, as follows. Class 1, affected individuals and normal spouses (penetrance = 1); class 2, radiologically normal individuals at 50% prior risk (penetrance = 0.8); class 3, individuals at 25% prior risk, normal by history (penetrance = 0.5). The affection status of individual IV-5 in family 2 was classed as unknown. Two-point lod scores were calculated by using MLINK of the FASTLINK package version 4.0P (ref. 20) at the UK HGMP-RC (http://www.hgmp.mrc.ac.uk). 2 Supplementary Information Bioinformatics and physical map construction. The GCG package was used for routine DNA and protein analysis. BLAST searches were carried out locally (http://www.molbiol.ox.ac.uk) or at NCBI (http://www.ncbi.nlm.nih.gov). An integrating script performing multiple BLAST queries and running several exon prediction programs simultaneously (J. Peden, unpublished), aided gene identification in genomic sequences. Protein databases, interrogated to find conserved elements, included InterPro (http://www.ebi.ac.uk/interpro) and Pfam (http://www.sanger.ac.uk/Software/Pfam/index.shtml). A publicly available resource at Southwestern Medical Center listing genomic clones covering the EXT2 region (Website discontinued) served as a starting point for map construction. We assembled PAC clones21 189f14 from RPCI-1 and 366b4, 368b4, 404m15, 511i10 and 526d1 from RPCI-3 (Research Genetics) into a contig using the existing framework of EXT2 exons22, markers D11S1393, D11S903 and D11S2095, and end-fragment isolation. The order of markers D11S1393 and D11S578 relative to EXT2 is based on the complete sequence of PAC clone 404m15 and contrasts with a former mapping4. Cosmid cSRL101h11 (ref. 3) and the RPCI-11 sequence-sampled BAC clones 70a24 and 706a13 were identified through BLAST searches as matches against PAC 404m15 and EXT2 exons respectively. We identified three putative genes between D11S1393 and EXT2: a gene of unknown function represented by the Unigene cluster Hs. 98649, a human homologue of a Fugu rubripes gene with significant similarity to the 1-aminocyclopropane-1-carboxylate synthase family from plants23 and a paralogue or pseudogene related to the latter. The localization of D11S2095 and size estimates for ALX4 introns 2 and 3 were based on long-range PCR of PAC clones 526d1 and 511i10 using the Expand Long Template PCR System (Roche). The mapping of MluI and AscI sites within exon 1, 3 Supplementary Information followed by inspection of genomic sequences flanking exons 1 and 2, enabled us to deduce the size of intron 1. The relationship of the ALX4 and EXT2 transcription units was corroborated by XhoI digestion of clone 526d1: probes corresponding to the terminal exons of each gene and D11S2095 all detect the same ~18 kb fragment. The 3´ end of ALX4 was further localised by identification of two ESTs (GenBank AW071529 and AW613995) containing a predicted poly(A) signal and mapping ~4 kb downstream from the termination codon. Molecular analysis of ALX4. Alignments with mouse Alx4 cDNA and the presence of canonical splice junctions demarcated human ALX4 exons in genomic sequences. We demonstrated the mutation in family 2 by amplification and sequencing of a 651 bp exon 1 product with the primer pairs 5´–GCAAGGAGTGCACAGCCACAGC–3´ and 5´–ACCAGTTTCAAGGGATGCGGAAG–3´; and the mutations in families 1, 3 and 4 by amplification of a 417 bp exon 2 product with the primer pairs 5´– CCCCCTGACATTCCCCTTCTCTT–3´ and 5´– GCTTTACCAGCCTCACTCCCAGGT–3´. A full list of primers is provided in Table 1. Uniform PCR conditions comprised an ammonium buffer (50 mM Tris HCl pH 9.2, 16 mM (NH4)2SO4, 2.25 mM MgCl2, 0.5% Tween 20, 10% dimethylsulfoxide) and annealing at 60 oC. SSCP analysis was carried out on the PCR products as described24 and sequencing was performed using the BigDye Terminator Cycle Sequencing kit (Perkin-Elmer) on the ABI 377 Sequencer. Restriction enzymes for mutation confirmation (loss of PvuII site in family 1, creation of BfaI site in family 2 and loss of MspI site in families 3 and 4) were obtained from New England Biolabs. Appropriate allele-specific oligonucleotides detected none of the mutations in a control panel of 48 unrelated north Europeans. We identified eight additional single nucleotide variants that 4 Supplementary Information we considered to be polymorphisms. The more common allele is listed first with its frequency in the same control panel in brackets: 104G>C encoding a R35T substitution (0.52), 304C>T encoding a P102S substitution (0.57), 594C>A (>0.97), 729G>A (0.92), 778-11G>A (0.94), 882C>T (0.93), 1074C>T (0.70), 1464C>T (0.75). The mutated alleles in families 3 and 4 exhibited different haplotypes for these polymorphisms, indicating that the two families are unrelated. cDNA isolation and Northern blot analysis. We reverse-transcribed total RNA from a normal human fibroblast cell line using M-MLV RT (Promega) and random hexamers. Amplification of ALX4 from cDNA employed the conditions and exon 1 forward primer given above, with the reverse primer 5´–ACCTGGCTTTCTCCACTGCCTGT–3´ from exon 4. This yielded a specific product of ~1.6 kb after 40 cycles which was directly sequenced. A Real Human Fetal mRNA Blot (Invitrogen) was hybridized in Ultrahyb buffer (Ambion) with an 32P-dCTP labelled PCR fragment from exon 4 of ALX4 (Megaprime DNA labelling system, Amersham Pharmacia) and washed under high stringency. An ALX4 transcript was also detected in a human fetal liver mRNA preparation (Clontech). In situ hybridization of mouse skull sections. In situ hybridization analysis of normal mouse embryos using Alx4 (ref. 9) and Spp1 probes was carried out as described15. Whole mount analysis with antisense probes at E14, E16 and E18 showed no visible staining (by comparison, the appearance at E10.5 was similar to that in ref. 9). Sections were cut from fresh frozen tissue, acetylated, fixed and prehybridized. Hybridization was carried out overnight using digoxygenin labelled probes and immunodetection was performed using BCIP. The equivalent sense probes gave no significant staining. 5 Supplementary Information Additionally, expression was observed in the mesenchymal tissue of early hair follicles and in the meninges, including the falx cerebri and tentorium cerebelli. References 16. Preis, S., Engelbrecht, V. & Lenard, H.-G. Aplasia cutis congenita and enlarged parietal foramina (Catlin marks) in a family. Acta Paediatr. 84, 701–702 (1995). 17. Wuyts, W. et al. Identification of mutations in the MSX2 homeobox gene in families affected with foramina parietalia permagna. Hum. Mol. Genet. 9, 1251– 1255 (2000). 18. Schmidt-Wittkamp, E. & Christians, H. Die Lückenbildungen der Scheitelbeine. Beobachtungen an 75 Mitgliedern einer Sippe mit gehäuftem Vorkommen von Foramina parietalia permagna. Fortschr. Geb. Rontgenstr. Nuklearmed. 113, 29– 38 (1970). 19. Dib, C. et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380, 152–154 (1996). 20. Cottingham, R.W.J., Idury, R.M. & Schaffer, A.A. (1993) Faster sequential genetic linkage computations. Am. J. Hum. Genet. 53, 252–263 (1993). 21. Ioannou, P.A. & de Jong, P.J. Construction of bacterial artificial chromosome libraries using the modified P1 (PAC) system. in Current Protocols in Human Genetics (eds Dracopoli, N.C. et al.) 5.15.1–5.15.24 (Wiley, New York, 1996). 22. Clines, G.A., Ashley, J.A., Shah S. & Lovett, M. The structure of the human multiple Exostosis 2 gene and characterization of homologs in mouse and Caenorhabditis elegans. Genome Res. 7, 359–367 (1997). 6 Supplementary Information 23. Peixoto, B.R., Mikawa, Y. & Brenner, S. Characterization of the recombinase activating gene-1 and 2 locus in the Japanese pufferfish, Fugu rubripes. Gene 246, 275–283 (2000). 24. Hayashu, K., Kukita, Y., Inazuka, M. & Tahira T. Single-strand conformation polymorphism analysis. in Mutation Detection A Practical Approach (eds Cotton, R.G.H., Edkins, E. & Forrest, S.) 8–24 (IRL Press, Oxford, 1998). 7 Supplementary Information Tables Table 1 Oligonucleotides used in characterisation of ALX4 and identification of mutations and polymorphisms PCR and Sequencing Primers Oligonucleotide Sequence Position & Orientation 5´–GCAAGGAGTGCACAGCCACAGC–3´ Exon 1, 5´ UTR; forward 5´–TAACAAGTTCCAGCCCCAGTCGTC–3´ Exon 1, coding region; forwarda 5´–CGCTGCAAGTAAAGATGCGGTTG–3´ Exon 1, coding region; reverse 5´–ACCAGTTTCAAGGGATGCGGAAG–3´ Intron 1; reverse 5´–CCCCCTGACATTCCCCTTCTCTT–3´ Intron 1; forward 5´–AGCTGGAGGAGCTGGAGAAGGTCT–3´ Exon 2; forward 5´–GTCTGTCCTCATGGCCAGCTGTT–3´ Exon 2; reverse 5´–GCTTTACCAGCCTCACTCCCAGGT–3´ Intron 2; reverse 5´–GGGAACAGTTTGCACTGCCTGAA–3´ Intron 2; forward 5´–CTCCTCCAAGGGGCTCATTCTCA–3´ Intron 3; reverse 5´–GAGCCCCTTCCACACCACACCT–3´ Intron 3; forward 5´–GCCTCAATGGCTACGAGCTCAACG–3´ Exon 4, coding region; forward 5´–ACTGTGCTCCTTGGCCTTCATGC–3´ Exon 4, coding region; reverse 5´–ACCTGGCTTTCTCCACTGCCTGT–3´ Exon 4, 3´ UTR; reverse Mutation and SNP detection ASOs Oligonucleotide sequence Variant detectedb 5´–CCTTTTACGGCATTT–3´ 104G>C SNP 8 Supplementary Information 5´–TGCGGCGACGGCTGG–3´ 304C>T SNP 5´–CCTTCCTAGAGTTTG–3´ 418C>T mutation 5´–CAGACCTACCCAGCC–3´ 594C>A SNP 5´–CGGAACCAGACCACC–3´ 653G>A mutation 5´–TGTATGCACGGGAAC–3´ 729G>A SNP 5´–GCCAGCTATTCCCGC–3´ 736C>T mutation 5´–CTGTTTCACGTCTTG–3´ 778-11G>A SNP 5´–GGGTGAGAAGGGGCA–3´ 882C>T SNP 5´–GGCCCACATGACTGC–3´ 1074C>T SNP 5´–TGTGGCCAGGAGCAG–3´ 1464C>T SNP a Further sequence analysis showed that this primer contains a mismatch (at italicized T) with respect to the usual wild type sequence. b Variant nucleotide is underlined. 9