Supplementary Materials - Springer Static Content Server
advertisement

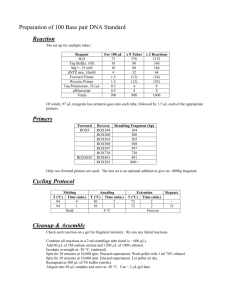
Supplementary Materials Generation of cell lines: PBMCs were prepared from available patients (1a and 6), parents and unrelated normal controls. Cells, after washing, were cultured with Epstein-Barr virus (EBV), PHA, and cyclosporine in RPMI complete media for 3- 4 weeks and frozen until ready to use. Genomic DNA amplifications were performed by touchdown PCR. Annealing temperatures ranged from 55 to 68°C. PCR products were purified by using the AgencourtR AMPureR XP PCR Purification Kit (Beckman Coulter, Beverly, Massachusetts), and sequenced with the BigDyeR Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, California) using the M13 Forward primer. After the sequencing reaction, the DNA was purified using the AgencourtR CleanSeqR Kit (Beckman Coulter, Beverly, Massachusetts) and run on a 3730xl DNA Analyzer (Applied Biosystems, Foster City, California). All kits were used in accordance with the manufacturer’s instructions. Sequencing data were analyzed for mutation detection using SeqMan II software (DNA Star Inc., Madison, WI). Samples with mutations were confirmed by sequencing the reverse strand using M13 Reverse primer. Once a mutation was found, one parent’s or both parent’s DNA, if available, were sequenced to determine their heterozygote (carrier) status for the same mutations as their children siblings. To rule out the possibility that the mutations detected were unreported normal variations within the Saudi population, 250 DNA samples from normal individuals obtained from the blood bank of the King Faisal Specialist Hospital and Research Centre were sequenced. Reverse transcription (RT)-PCR was performed with the following primers: Primers 1: 5’-CAGGGGAGTCAGCAGAGG-3’ (forw); 5’-ACAGGCCTCACTCGTACAGT-3’ (rev). Primers 2: 5’-ACCTAGGGCTTCGGGTC-3’ (forw); 5’ACATCAGTCTTGTTTGTGCC-3’ (rev). Primers 3: 5’AAATGTCACCCTTGGACAAG-3’ (forw); 5’-CTCTGGCCAACTGCCTG-3’ (rev). Primer 4: 5’-GCATCTCAGTGCAGGAGAGA-3’ (forw); 5’AACAATGCTGCAATGGGC-3’ (rev). Primer 5: 5’-CATCATGGTGGCTTCCCT-3’ (forw); 5’-AGACCCTTCTTCCCCACC 3’ (rev). In addition, M13 sequences were attached to the 5’ end of each primer to allow forward and reverse sequencing. The PCR conditions were 1 cycle at 94°C for 3 min, 35 cycles at 94°C for 30 s, at 55°C for 30 s, and at 72°C for 30 s, and 1 cycle at 72°C for 3 min. To assess exon 3 skipping, RT-PCR was performed in a patient and one family member of that patient from exon 1 to exon 4 using primers 5’-CAGGGGAGTCAGCAGAGG-3’ (forw) and 5’AACAATGCTGCAATGGGC-3’ (rev), from exon 2 to exon 4 using primers 5’ACCTAGGGCTTCGGGTC-3’ (forw) and 5’-AACAATGCTGCAATGGGC-3’ (rev), and from exon 3 to exon 4 using primers 5’-AAATGTCACCCTTGGACAAG-3’ (forw) and 5’-AACAATGCTGCAATGGGC-3’ (rev). Western blot analysis was performed as follows. Briefly, Laemmli sample buffer (BioRad, USA) supplemented with 1/20 volume of Beta-mercaptoethanol (βME; Sigma, USA) was added to the protein samples at a 1:1 ratio and boiled for 5 minutes. Proteins were then separated on a 12% ready-made Criterion XT Bis-Tris gel (BioRad, USA) and transferred to a Trans-Blot Transfer medium 0.2 m nitrocellulose membrane (BioRad, USA). Taking advantage of the difference in size between CD40 (50 kD) and STAT5 (90 kD), filters were cut horizontally in two halves and the bottom half was probed with an affinity-purified rabbit polyclonal antibody to the carboxyl- (SC-975, Santa Cruz,CA, USA) which recognizes only the 40 kD variant and/or amino- (5853, Abcam) terminus of the CD40 protein. . The top half was probed with an affinitypurified rabbit polyclonal antibody against the C-terminus of the signal transducer and activator of transcription 5 (STAT5) protein (SC-835; Santa Cruz, USA) as a loading control. HRP-goat anti-rabbit antibody (65-6420, Invitrogen, USA) was used as a secondary antibody. The blots were developed using Super Signal West-Pico chemiluminescence (Pierce, USA). Supplementary Fig. 1: Flow cytometry analysis of CD40 expression on B cells for control compare to all 11 patients. Panels show double staining for PE anti-CD40 and APC, PerCP, or FITC anti-CD19. Supplementary Fig. 2: c. 256+2T>C mutation. Electrograms of the four family members, sequenced as indicated. The line along the electrograms is placed next to the mutation site. The arrow points to the mutation. P = patient. Supplementary Fig. 3: (a) The sequence shows the sequence of the cryptic splice site within exon 3. Below the sequence, the output of the splice prediction program is shown. (b) A sequence showing the result of the misspliced transcript and the expected protein sequence. (c) RT-PCR of two additional NCs showing the correctly spliced cDNA. Supplementary Fig. 4: T57M mutation: (a) Electrograms of the two family members, sequenced as indicated. P = patient. (b) Alignment of protein sequence of the indicated species around the mutated amino acid. The affected amino acid (T) is bolded, showing its conservation among the many species indicated. (c) Output of PolyPhen2 prediction. The output predicts a very high probability of the mutation being damaging, as indicated by a score of 1. Supplementary Table: in vitro T cell proliferation in response to PHA (cpm) Patient 1a 1b 2 3 4a 4b 4c 5a 5b 6 7 Patients PHA(cpm) NA 84,424 69498 115516 NA 70253 NA 92020 48649 NA 40391 Control –1 PHA(cpm) Control – 2 PHA(cpm) % of Control 142,755 104,435 138,510 140,082 123,703 138,510 60% 61% 84% 101,709 103,327 68% 97583 81310 101595 76070 92% 62% 126,609 169,539 27% ** 2.5×105 mononuclear cells were cultured in 0.5 mL of RPMI 1640 plus 10% AB serum for 3 days in the presence of PHA (10 μg/mL). The results of triplicate cultures are expressed as the mean counts per minute of [3H] thymidine incorporation (±1 SD) cpm (counts per minute).