Supplementary information - Springer Static Content Server
advertisement

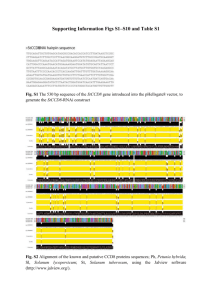
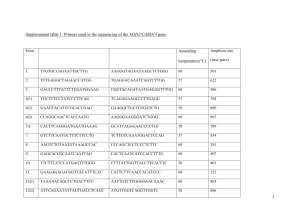
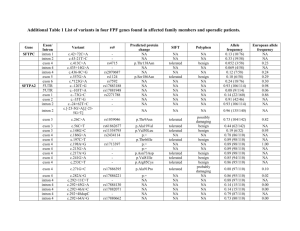
Supplementary information Figure S1 Alignment of the amino acid sequences deduced from the Aime-MHC-I genes. The boxed symbols represent types of intron 2 sequences, as shown in Figure S2. The underlined sequence shows the putatively classical but monomorphic Aime-F gene [1], and the light- and dark-grey regions indicate the two wild-specific haplotypes and two nonclassical loci uninvolved in this study, respectively. These five sequences were from GenBank (JX987008, JX987023, EU162656, EU162657 and EU162661). Dots indicate identities and gaps represent missing amino acids to the first sequence. Crosses represent putative antigen-binding sites, corresponding to the method of Bjorkman et al. [2]. 1 2 Figure S2 Inter-haplotype sharing of intron 2 sequences (A) and nucleotide sequence alignment (B) of the Aime-MHC-I genes. The black and gray lines represent classical and nonclassical loci, respectively (A). Dots indicate identities and gaps represent missing amino acids to the first sequence (B). The shaded intons were from GenBank (EU162656, EU162657 and EU162661). The Aime-C*09 (JX987008) and Aime-L*07 (JX987023) were not detected in this study but shared the intronic sequences with other haplotypes. 3 Table S1 Haplotypic frequencies and heterozygosities of the three MHC class I loci. Haplotype Aime-C Aime-I Aime-L Mean 01 0.011 0.156 0.089 / 02 0.344 0.522 0.433 / 03 0.100 0.089 0.122 / 04 0.033 0.144 0.089 / 05 0.222 0.011 0.022 / 06 0.267 0.044 0.244 / 07 0.011 0.033 / / 08 0.011 / / / Na 8 7 6 7 HO 0.667* 0.667* 0.644* 0.659 HE 0.758 0.679 0.729 0.722 Note: Na is the number of haplotypes. Asterisks indicate significant deviation from Hardy-Weinberg equilibrium (P < 0.05). The figures in bold represent the predominant haplotype for each locus. 4 Table S2 Nonsynonymous and synonymous substitutions at selected sites (positive or purifying) within the three Aime-MHC-I genes using REL methods. Locus Aime-C Aime-I Aime-L Codon E[dS] E[dN] Normalized Posterior Bayes E[dN-dS ] probability factor 70+ 0.53 4.78 4.25 0.97 73.67 73+ 0.18 1.42 1.24 0.99 345.34 76+ 0.43 1.42 0.99 0.97 55.38 80+ 0.42 1.41 0.99 0.97 55.78 83 0.34 1.43 1.09 0.97 82.32 94 0.43 1.42 0.99 0.96 55.23 118* 8.83 0.22 -8.61 0.99 205.39 119* 8.88 0.19 -8.69 0.99 191.40 152+ 0.91 15.52 14.61 0.99 281.12 63+ 0.98 1.77 0.79 0.95 84.41 66+ 0.77 1.77 1.00 0.98 165.08 70+ 0.81 1.75 0.94 0.96 103.89 73+ 0.55 1.77 1.22 1.00 7164.58 74+ 0.87 1.76 0.88 0.96 91.33 81+ 1.04 1.77 0.73 0.94 71.34 97+ 0.78 1.76 0.98 0.97 130.06 103 0.62 1.76 1.14 0.98 272.08 114+ 0.73 1.77 1.04 0.98 203.41 152+ 0.90 1.77 0.87 0.96 109.15 163+ 0.92 1.77 0.85 0.96 99.91 23 1 3.50 2.50 0.90 59.16 24+ 1 3.54 2.54 0.91 66.12 116+ 1 3.81 2.81 0.99 483.55 156+ 1 3.78 2.78 0.98 284.68 160 1 3.78 2.78 0.98 281.97 5 Notes: E[dS] and E[dN] indicate posterior means of the synonymous and nonsynonymous substitution rates at each site. E[dN-dS] indicates the posterior mean of the dN-dS difference. The asterisks and crosses represent the purifying selection and antigen-binding sites, respectively. 6 Table S3 Nonsynonymous and synonymous substitutions within the three Aime-MHC-I genes using MEGA 4.0. Locus Aime-C Aime-I Aime-L Position N dN dS dN / dS P ABS 57 0.104 (0.028) 0.034 (0.018) 3.059 0.004 Non-ABS 125 0.007 (0.004) 0.024 (0.013) 0.292 1.000 All 182 0.036 (0.008) 0.027 (0.010) 1.333 0.240 ABS 57 0.101 (0.025) 0.066 (0.023) 1.530 0.142 Non-ABS 125 0.008 (0.003) 0.037 (0.014) 0.216 1.000 All 182 0.036 (0.008) 0.046 (0.011) 0.783 1.000 ABS 57 0.045 (0.016) 0.017 (0.011) 2.647 0.025 Non-ABS 125 0.010 (0.004) 0.030 (0.011) 0.333 1.000 All 182 0.021 (0.006) 0.026 (0.008) 0.808 1.000 Notes: Standard errors (in parentheses) were obtained through 1000 bootstrap replicates. N is the number of codons, and P-value shows statistical significance of differences between dN and dS. 7 Data S1 Materials and Methods Sampling We collected 46 blood samples during routine medical examination of giant pandas in the China Research and Conservation Center for the Giant Panda (Wolong). Genomic DNA was extracted using the conventional phenol-chloroform method [3]. Amplification and genotyping The exons 2 and 3 of MHC class I genes encode domains responsible for antigen presentation. Thus, we amplified the exon 2–intron 2–exon 3 using the locus-specific primer pairs published by Zhu et al. [1]. We cloned the PCR products and amplified the resultant clones using the universal nested primers for separate exon 2 and exon 3 of the Aime-MHC-I genes (Table S4). The nested PCR fragments of exons 2 and 3 were subjected to single-strand conformation polymorphism (SSCP) analysis. We sequenced the positive clones which have different combinations of exon 2 and exon 3 banding patterns. Each exon 2–intron 2–exon 3 haplotype was confirmed by sequencing at least three independent clones. The PCR and SSCP conditions were described in Zhu et al. [1]. Data analysis We calculated haplotypic frequencies and tested deviations from Hardy-Weinberg equilibrium for each locus using Genepop4.0 [4]. Observed (HO) and expected (HE) heterozygosities were estimated in Arlequin3.1 [5]. All the sequences were edited and aligned using the DNASTAR software package (DNASTAR, Inc.). We checked for evidence of recombination on the exon 2, 8 intron 2, and exon 3 fragments using the maximum likelihood-based GARD (genetic algorithm for recombination detection.) [6] and chi-square-based MaxChi2 (modified maximum chi-square) [7] methods. A GARD analysis was conducted through a web-based routine (http://www.datamonkey.org/GARD/) while MaxChi2 were implemented in RDP 3.44 [8]. We tested positive selection at the Aime-MHC-I genes using a Random Effects Likelihood (REL) method implemented in the HyPhy Package (http://www.datamonkey.org/) [9]. REL adopts a maximum likelihood method to compute rates of nonsynonymous (dN) and synonymous (dS) substitutions for each codon [10]. Empirical Bayes analysis was then used to determine whether there was evidence of positive selection (dN > dS) or purifying selection (dN < dS) when Bayes factors are larger than 50. Since recombination is known to seriously overestimate positive selection [11, 12], we detected recombination events in our datasets using the GARD method described above prior to implementing REL. The HyPhy Package can detect evidence of selection in the presence of recombination. The dS/dN ratios were also calculated using MEGA 4.0 [13] and standard errors were estimated through 1000 bootstrap replicates. To test for signatures of balancing selection at the three Aime-MHC-I genes, we used a Z-test as implemented in MEGA 4.0 to compare the rates of synonymous and nonsynonymous substitutions at all sites, including antigen binding sites (ABSs) and non-ABSs. ABSs were identified by comparison to human sequences [2]. The number of nucleotide substitutions in the intron 2 (d) and the number of 9 synonymous substitutions in the exons 2 and 3 (dS) were estimated using a Kimura-2p method in K-estimator [14]. We plotted mean d and dS against nucleotide position using a sliding window size of 15 base pairs and a step size of 3 base pairs. References for Supplementary Information 1. Zhu Y, Wan QH, Yu B, et al. BMC Evol Biol 2013; 13:227. 2. Bjorkman PJ, Saper MA, Samraoui B, et al. Nat Immunol 1987; 329:512-518. 3. Sambrook J, Russell DW (2001) Molecular Cloning: A Laboratory Manual, 3rd edn. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York). 4. Rousset F. Mol Ecol Resour 2008; 8:103-106. 5. Excoffier, Laval LG, Schneider S. Evol Bioinform Online 2005; 1:47-50. 6. Pond SLK, Posada D, Gravenor MB, et al. Mol Biol Evol 2006; 23:1891-1901. 7. Maynard-Smith J. J Mol Evol 1992; 34:126-129. 8. Heath L, Van Der Walt E, Varsani A, et al. J Virol 2006; 80:11827-11832. 9. Pond SLK, Frost SDW. Bioinformatics 2005; 21:2531-2533. 10. Pond SLK, Frost SDW. Mol Biol Evol 2005; 22:1208-1222. 11. Anisimova M, Nielsen R, Yang Z. Genetics 2003; 164:1229-1236. 12. Richman AD, Herrera LG, Nash D, et al. Genet Res 2003; 82:89-100. 13. Tamura K, Dudley J, Nei M, et al. Mol Biol Evol 2007; 24:1596-1599. 14. Comeron JM. Bioinformatics 1999; 15:763-764. 10 Table S4 Locus-specific primers of the three polymorphic Aime-MHC-I genes. The primer sequences were published by Zhu et al. [1]. Locus Primer Primer sequence (5’-3’) name Aime- C Aime-I Aime-L Aime-C/I/ 1611E21F a GCTCTCCCCCACTCAGTA 1611E33R a TGCAGGTCTAAGAGGGAGAGCGCT 128E22F a CCTGCTCTCCCCAACGCG 128E32R a GTCCGGGGTTTCTGAAGAAGAACG 1300E22F a GGGAGAAGGGTCGGGCGGGAC 1300E31R a GTCGGGGGTTTCTGAAGAAGAATC C1E2B1 b TCAGCCCCTCCGCGCCCGCAG C1E2B2 b GACCCGGGCCGCGTCGCTCAC C1E3B1 b TCGCCTCCTGTCGGGCGGGGCCAG C1E3B2 b AGCCAGCCCCAGCGGAGGGG Size Ta (bp) (C) 1255 60.0 1467 60.0 1354 60.0 312 66.3 349 65.6 LE2 Aime-C/I/ LE3 Notes: The primer pairs (a) were used to amplify the long fragment of exon 2–intron 2–exon 3, and the primer pairs (b) were then adopted for the subsequent nested PCR to get separate exon 2 and exon 3 fragments for single-strand conformation polymorphism analysis. 11