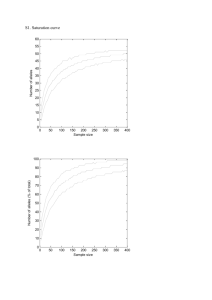
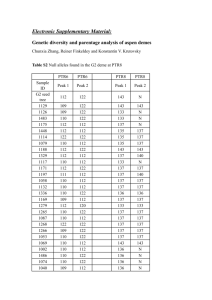
Supplementary Information (doc 39K)
advertisement
")
1 SUPPLEMENTARY METHODS 2 Within group relatedness estimation 3 Presence of related individuals in the population can affect estimates of 4 population differentiation. Pairwise relatedness between the samples was 5 estimated with the Triadic likelihood estimator (Wang, 2007) implemented in 6 the program COANCESTRY. The TrioML method uses a third individual as 7 reference to estimate the relatedness between two other individuals, thus 8 reducing the chance of alleles identical in state being mistaken for alleles 9 identical by descent. This method also allows for inbreeding and genotyping 10 errors, and thus is suitable for most natural population studies. We estimated 11 the average within group relatedness for each population of both the species. 12 Test for neutrality of loci 13 We tested if our microsatellite loci are selectively neutral using the 14 method described in Beaumont & Balding (2004) as implemented in the 15 program Bayescan v 2.1 (Foll & Gaggiotti, 2008) which uses outlier analysis to 16 identify candidate loci under selection. As it uses the Bayesian approach, it 17 incorporates uncertainty in allele frequencies due to small sample sizes. We ran 18 the dataset for each species separately for 10,000 simulations with a thinning 19 interval of 50 and a burn-in of 5000. Bayescan reports posterior probability for 20 a model incorporating natural selection and introduces a criterion for model 21 choice similar to Bayes factor called Posterior Odds (PO) which is a ratio of the 22 posterior probabilities. Low values of log10(PO) suggest no evidence for 23 selection. 24 Calculation of genotyping errors 25 We repeated PCR amplification and genotyping for 94 individuals of R. 26 satarae and 27 individuals of R. rattus at least twice to quantify genotyping 27 errors using the program GIMLET. We quantified four types of errors including 28 allelic dropout and false alleles. The error rates for R. satarae were slightly 29 higher than that for R. rattus (1% and 0.5% respectively). 30 Detection of null alleles and estimation of null allele frequencies 31 Since the same genetic markers were used on two different species, presence of 32 null alleles is possible and could possibly explain observed heterozygote deficit. 33 We evaluated this with the program MICROCHECKER (van Oosterhout et al. 34 2004). Further, we estimated the frequency of null alleles for each locus and 35 population using the Expectation Maximisation algorithm (Dempster et al. 36 1977) implemented in the software FreeNA (Chapuis & Estoup 2007). 37 38 39 REFERENCES 40 41 42 43 Beaumont, M. A., & Balding, D. J. (2004). Identifying adaptive genetic divergence among populations from genome scans. Molecular Ecology, 13(4), 969–980. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15012769 44 45 46 Chapuis, M.-P., & Estoup, A. (2007). Microsatellite null alleles and estimation of population differentiation. Molecular Biology and Evolution, 24(3), 621–31. doi:10.1093/molbev/msl191 47 48 49 Foll, M., & Gaggiotti, O. (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics, 180(2), 977–993. doi:10.1534/genetics.108.092221 50 51 52 Wang, J. (2007). Triadic IBD coefficients and applications to estimating pairwise relatedness. Genetical Research, 89(3), 135–53. doi:10.1017/S0016672307008798 53