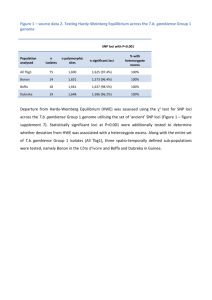
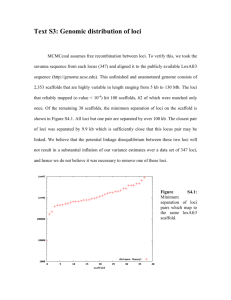
click here and type title
advertisement

International Biometric Society APPROXIMATE BAYESIAN COMPUTATION APPLIED TO NONINVASIVE DNA-BASED POPULATION ESTIMATION Bruce A Craig, Purdue University Shannon Knapp, New Mexico State University DNA from non-invasive sources (primarily hair and scat) is now commonly used as tags for mark-recapture population estimation. These DNA sources are particularly useful for species that are secretive, endangered, or sparsely distributed. Unfortunately, these non-invasive sources are prone to genotyping errors, corrupting the true “mark” and resulting in “ghosts” – what appear to be additional individuals. Additionally, as with any source of DNA, using insufficient genetic information (i.e., too few loci or insufficiently variable loci) can result in the “shadow effect” wherein two individuals share the same “mark.” A number of methods, laboratory-based and statistical, have been proposed to address both these problems. Of these methods, most require replicate amplification of individual samples, an undesirable property as this incurs additional cost and effort. For this talk, we focus on the case of a small population size and small sample size (i.e., secretive or sparsely distributed species). In this situation, previous results have shown asymptotic-based confidence intervals perform poorly. While a fully Bayesian analysis would be desirable, the likelihood of the data (a collection of observed genotypes, with errors) is computationally very costly, as it involves not only the true population size, but the capture history (i.e., all possible ways all individuals could be caught one or more times) and the true genotypes for those individuals caught. To get around this, we propose using Approximate Bayesian Computation (ABC) methods. ABC works by simulating data for a range of parameter values (here, the parameter of interest is N, the population size) and then keeping only those simulations (and thus the proposed N) for which the distance from the simulated data to the observed data is within some threshold. These methods have been used for problems in population genetics, specifically for estimating coalescence times (time to most recent common ancestor), but have not been used for population estimation from noninvasive DNA. Through simulation, we explore a number of candidate measures of distance between the observed and simulated data (and combinations thereof) including: the number of pairs of samples that were a perfect match at all loci, the number of pairs of samples that were a potential match at all loci allowing for the most common forms of genotyping errors, the number of pairs of scats that were perfect matches at 0, 1, . . . , L loci, and the probability of getting a pair of observed genotypes given the samples came from the same individual for all pairs of samples. For each combination of distance measures, the distribution of retained N’s forms the posterior distribution for N – yielding both an estimate and a credible interval. We summarize the properties of these intervals and demonstrate that for well-chosen distance measures, these ABC methods outperform the asymptotic-based intervals. International Biometric Conference, Florence, ITALY, 6 – 11 July 2014