슬라이드 1
advertisement

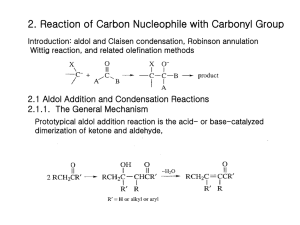
2. Reaction of Carbon Nucleophile with Carbonyl Group Introduction: aldol and Claisen condensation, Robinson annulation Wittig reaction, and related olefination methods 2.1 Aldol Addition and Condensation Reactions 2.1.1. The General Mechanism Prototypical aldol addition reaction is the acid- or base-catalyzed dimerization of ketone and aldehyde, The equilibrium constant for the dehydration phase is usually favorable, because of the conjugated a,b-unsaturated carbonyl system that is formed. 2.1.2 Mixed Aldol condensation with Aromatic Aldehyde One of the most general mixed aldol condensation inovolves the use of aromatic aldehyde with alkyl ketones or aldehyde. Non-enolizable Claisen-Schmidt Condensation Pronounced preference for the formation of a trans double bond in the Claisen-Schmidt condensation of methyl ketones. Base-catalyzed dehydration is slow relative to the reverse of the addition phase for the branched-chain isomer. In base, the straight-chain ketol is the only intermediate which is dehydrated. The branched chain ketol reverts to starting material. Under acid condition, both intermediates are dehydrated, however, the branched-chain ketol is formed most rapidly, because of the preference for acid-catalyzed enolization to give the more substituted enol. Under acid condition, Both intermediates are dehydrated, however, the branched-chain ketol is formed most rapidly, because of the preference for acid-catalyzed enolization to give the more substituted enol forms rapidly major Base catalysis favors reaction at a methyl position over a methylene group, whereas acid catalysis gives the opposite preference. 2.1.3. Control of Regiochemistry and Stereochemistry of Mixed Aldol Reactions of Aliphatic Aldehyde and Ketones 2.1.3.1. Lithium Enolates Kinetic controlled conditions Directed Aldol Reaction Cyclic Transition State E-enolate Anti-ketol The enolate formed from 2,2-dimethyl-3-pentanone under kinetically controlled conditions is the Z-isomer. Reaction with benzaldehyde gives syn aldol. When alkyl substituent of ketone is bulky, Z-enolate is formed. And synaldol product is formed. Order: t-butyl>i-propyl>ethyl The enolate of cyclohexanone reacts with benzaldehyde are necessarily E-isomers. Anti-isomer is major. Because the aldol reaction is reversible, it is possible to adjust reaction conditions so that the two stereoisomeric aldol products equilibriate. 1) Z-enolate syn aldol; E-enolate anti aldol 2) When the enolate has no bulky substituents, stereselectivity is low 3) Z-enolates are more stereoselctive than E-enolates. Ref. Table 2.1 For synthetic efficiency, it is useful to add MgBr2. The greater stability of the anti-isomer is attributed to the pseudoequitorial position of the methyl group in the chair-like chelate. With larger substituent groups, the thermodynamic preference for the anti-isomer is still greater. Ketones with one tertiary alkyl substituent give mainly the Z-enolate. However, less highly substituted ketones usually give mixtures of E- and ZEnolates. Control of stereochemistry of aldol reaction (1) Control of enolate stereochemistry (2) enhancement of the stereoselectivity in the addition step. For simple ester, the E-enolate is preferred under kinetic conditions using a strong base such as LDA in THF. But Inclusion of a strong cation sovating co-solvent, such as HMPA favors the Z-enolate. With LDA/THF conditions, cyclic transition state, an open transition state in the presence of an aprotic dipolar solvent If R= bulky, selectivity is increased Simple alkyl esters show rather low stereoselectivity. Highly hindered esters provide the anti-stereoisomers. See Table 2.2. HMPA Z-enolate Syn-major a-alkoxy ester: higher stereoselectivity in some cases: it can be explained In terms of a chelated ester enolate. The aldehyde R group avoids being between the a-alkoxy and the methyl group in the ester enolate. When the ester alkyl group R becomes very bulky, the stereoselectivity is reversed. The allylic stabilization of the c-deprotonation product can lead to kinetic selectivity in the deprotomation. 2.1.3.2. Boron Enolates The stereoselctivity is higher than for lithium enolates, since the O-B bond distances are shorter than the O-Li bond in the lithium enolates, and this leads to a more compact transition state. Trifluoromethanesulfonate = triflate Z-isomer Syn-isomer E-boron enolate Anti-isomer Use of boron triflates with a more hindered amine favors the Z-enolate. The E-boron enolates of some ketone can be preferentially obtained with the use of dialkylboron chlorides. Cyclic mechanism for hydride transfer E-boron enolate Z-enolate Anti-aldol product Boron enolates parallel lithium enolates in their stereoselectivity but show enhanced stereoselectivity. (ref. table 2.3) 2.1.3.3. Titanium, Tin, Zirconium Enolates: intermediate between Li+ and covalent boron enolate. Z-enolate Cyclic transition state N-acyloxazolidinone Syn-aldol catalytic Tin enolates N-acylthiazolinethiones E-enolates Syn-selective (Cp)2ZrCl2 with lithium enolate Addition of silyl enol ethers can be catalyzed by (Cp)2Zr2+ species. The order of stereoselectivity is Bu2B>(Cp)2Zr>Li. These results are consistent With reactions proceeding through a cyclic transition state. 2.1.3.4 The Mukaiyama Reaction: Lewis-acid-catalyzed aldol addition reactions of enol derivatives. Not a strong enough nucleophile, but with Lewis acid the reaction proceeds through an acyclic transition state. For a-substituted aldehyde show a preference for a syn relationship between the a-substituent and hydroxy group. This is consistence with a Felkin-Ahn Transition state. 2.1.3.5. Control of Enantioselectivity The combined interactions of chiral centers in both the aldehyde and the enolate determine the stereoselectivity. The result is called double stereodifferentiation. The oxazolinone substituents R’ direct the approach of the aldehyde. 2.1.4. Intramolecular Aldol Reaction and the Robinson Annulation Robinson Annulation is a procedure which construct a new 6-membered ring from a ketone. Originally thermodynamic controlled reaction is required. The role of the trimethylsilyl group is to stabilize the enol formed in the conjugate addition. The silyl group is then removed during the dehydration step. It can be used under aprotic conditions. The s-enantiomer of the product is obtained in high enantiomeric excess with L-proline,. L-proline participates in the proton-transfer step. 2.2. Addition reactions of Imines and Iminium Ions. The reactivity order is C=NR<C=O<[C=NR2]+<[C=OH]+. 2.2.1. the Mannich Reaction: the condensation of an enolizable carbonyl compound with an iminium ion. The reaction is usually limited to secondary amines, because dialkylation can occur with primary amines. The dialkylation reaction can be used in ring closure. Synthesis of Mannich base Bis(methylamino)methane N,N-Dimethylmethyleneammonium idode “Eschenmoser’s salt” Thermal elimination of the amines or the derived quaternary salts provides a-methylene carbonyl compounds. Vernolepin having antileukemia activity Tropinone, alkaloid tropine by Sir Rober Robinson in 1917 2.2.2. Amine-Catalyzed Condensation Reactions Amine and acid are required: mixed aldol followed by dehydration: catalyzed by amine and buffer system: Knoevenagel condensation. Malonic ester, cyanoacetic ester, cyanoamide are examples of compounds which undergo condensation reactions under Knoevenagel conditions. Nitroalkanes are also good nucleophilic reagent in which a hydrogens are deprotonated under weakly basic conditions. Secondary amine is used as catalysts, iminium ion is involved in addition step. Decarboxylative condensation is carried out in pyridine, which can not form an imine intermediate. concerted decarboxylation Enzyme-Metal Combo Catalysis as a New Chirotechnology 김만주 교수 포항공대 화학과 2.3. Acylation of Carbanions Ester self-condensation is Claisen Condensation. Most acidic species Final step drives the reaction to completion. When a-substituted ester are used, it do not condense under the normal reaction conditions. Very strong base converts the ester completely to its enolate.: sodium hydride Intramolecular version of ester condensation is called the Dieckmann condensation Because ester condensation is reversible, product structure is governed by thermodynamic control: The product is derived from the most stable enolate. Acylation of ester enolates can be carried out with more reactive acylating agents such as acid anhydrides and acyl chlorides: the reaction must be done in inert solvents to avoid solvolysis of the acylating reagent. N-methoxy-N-methylamides is also useful for acylation of ester enolates. Sometimes O-alkylation is problem, and magnesium enolates play an important role in C-acylation reaction. Acyl imidazolides are more reactive than esters but not as reactive as acyl halides 2.4 The Wittig and Related Reactions An ylide is a molecule that has a contributing Lewis structure with opposite charges on adjacent atoms, each of which has an octet of electrons. Phosporus ylides are stable, but usually quite reactive. Organolithium compounds Unstabilized ylides give predominantly the Z-alkene whearas stabilized ylides give mainly the E-alkene. Use of sodium amide or sodium hexamethyldisilylamide as bases gives higher selectivity for Z-alkenes than with alkyllithium reagent as base. The three phenyl substituents on phosphorus impose large steric demands which govern the formation of the diastereomeric adducts. Reactions of unstabilized phosphoranes are believed to proceed through an early transition state, and steric factors usually make such transition states selective for the Z-alkene. Z-formation mechansim Schlosser modification of the Wittig reaction: the reaction of unstabilized ylide with aldehyde can be induced to yield E-alkenes with high stereoselectivity. b-oxido ylide Syn-elimination E . V e d e js , e t a l J . Am . C h e m . S o c ., 1 9 7 3 , 9 5 , 5 7 7 8 R R P R R' O R 3P O C H C R R H H H R' The reaction of unstbilized ylide proceeds through early transition state. That of stabilized ylide proceeds through late transition state. R' R R C O H H P R R 3P = O C H R' R' + H R' S ch lo sser's m o d ificatio n : ad d itio n o f P h L i H 3C H cis R' + O =PPh3 H Li Ph 3 P + CH CH 3 H CH 3 H Ph 3 P R 'C H O LiO PhLi CH 3 Ph 3 P H LiO R' H 3C H O =PPh3 + H R' Ph 3 P LiO + R' CH 3 t-BuOH 1) HCHO o H H 2) 25 C R' tra n s H 3C H CH 2 OH R' Phosphonoacetate esters are used to prepare a,b-unsaturated esters: Wadsworth-Emmons reaction: usually lead to the E-isomer. b -C a ro te n e A rb u zo v R e a c tio n O (CH 3 O) 3 P CH 3 I + CH 3 O P CH OCH 3 3 I C 2H 5 (CH 3 O) 3 P + (CH 3 O) 2 C 2H 5I C 2H 5 (CH 3 O) 2 P O + CH 3 I P O CH 3 Three modified phosphonoacetate esters have been found to show selectivity for the Z-enoate product. Trifluoroethyl, phenyl, 2,6-difluorophenyl esters give good Z-stereoselectivity. Carbanions derived form phosphine oxide add to carbonyl compounds. The adducts are stable but undergo elimination to form alkenes on heating with a base such as sodium hydride.: Horner-Wittig reaction. Usually anti-adduct is the major product, so it is the Z-alkene which is favored. The syn adduct is most easily obtained by reduction of b-keto phosphine oxide. 2.5 Reactions of Carbonyl Compounds with a-trimethylsilylcarbanions b-Hydroxyalkyltrimethylsilanes are converted to alkenes in either acidic or basic solution. It begins with nucleophilic addition of an a-trimethylsilylsubstituted carbanion to an aldehyde or ketone (Peterson reaction). The separate elimination step is not necessary because fragmentation of the intermediate occurs spontaneously. The elimination reactions are anti under acidic conditions and syn under basic conditions: the result of a cyclic elimination mechanism under basic conditions, whereas an acyclic b-elimination under acidic conditions. The anti-elimination can also be achieved by converting the b-silyl alcohol to trifluoroacetate esters. 2.6 Sulfur Ylide and related Nucleophiles Sulfur ylides are prepared by deprotonation of the corresponding sulfonium salts. Phosphorus ylides + Ketone alkene Sulfonium or sulfoxonium ylides + ketone epoxide Intramolecular displacement Dimethylsulfonium metylide is less stable than dimethylsulfoxonium methylide, so it is generated and used at a low temperature. 2.7 Nucleophilic Addition-Cyclization Darzens Reaction: The first step is addition of the enolate of the a-halo ester to the carbonyl compound followed by intramolecular SN2 reaction. Trimethylsilyl epoxide can be also preapred by an addition-cyclization process.