Advanced Organic Chemistry
advertisement

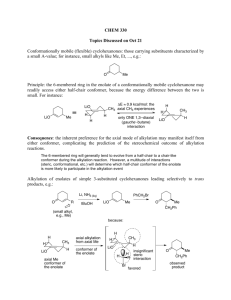
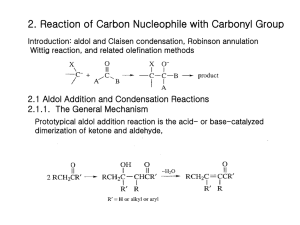
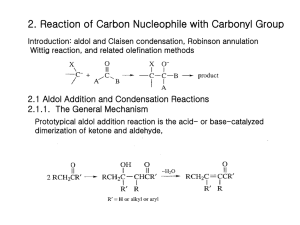
Advanced Organic Chemistry Part B: Reactions and Synthesis 4th Edition Francis A. Carey and Richard J. Sundberg Kluar Academic / Plenum Publishers Chapter 1. Alkylation of Nucleophilic Carbon Intermediates • Introduction C-C bond formation is the basis for the construction of the molecular frame work of organic molecules by synthesis. One of fundamental processes for C-C bond formation is a reaction between a nucleophilic carbon and an electrophilic one. Reaction of C-nucleophile (enolate ions, imine anions, enamines) with alkylating agents Crucial Factor for C-C bond formation by SN2 reaction • (1) the condition for generation of the carbon nucleophile • (2) the effect of the reaction conditions on the structure and reactivity of the nucleophile • (3) the regio- and stereoselectivity of the alkylation reaction • (4) the role of solvent, counterions, and other components of the reaction media that can influence the rate of competing reactions 1.1 Generation of Carbanions by Deprotonation • The rate of deprotonation and the stability of the resulting carbanion are enhanced by the presence of substituent groups that can stabilize negative charge. • Several typical examples of proton abstraction equilibria is shown in scheme 1.1 • Favorable equilibrium between a carbon acid and its carbanion will be established if the base which is used appears below the acid in the table 1.1 • An ordering of some important substituents with respect to their ability to stabilize carbanion can be established. NO2> COR>CN-CO2R>SOR>Ph-SR>H>R Strong base, but it is sufficiently bulky so as to be relatively nonnucleophilic. Lithium, sodium, potassium of hexamethyldisilazane, [(CH3)2Si]2NH Aprotic solvent: ether, tetrahydrofurane (THF), dimethoxyethane (DME) 1.2 Regioselectivity and Stereoselectivity in Enolate Formation Ideal conditions for kinetic control of enolate formation are those in which deprotonation is rapid, quantitative, and irreversible. Lithium is better counterion than sodium or potassium for regioselective generation of the kinetic enolate, since lithium maintains a tighter coordination at oxygen and reduces the rate of proton exchange. Aprotic solvents are essential because protic solvents permit enolate equilibrium by reversible protonation-deprotonation, which gives rise to the thermodynamically controlled enolate composition. Excess ketone also catalyzesthe eqiulibriation by proton exchange. Conditions of kinetic control usually favor the less substituted enolate. At equilibrium, thermodynamic controlled conditions, the more substituted enolate is usually the dominat species. 1.3 Other Means of Generating Enolates Driving force: very strong Si-F bond energy (142 kcal/mol) 1.4 Alkylation of Enolates SN2 Displacement Primary halide, sulfonates, allyl benzyl > sec halide >> t-alkyl halide (only elimination) The rate of cyclization: intramolecular cyclization for w-haloalkyl malonate esters 3 : 4 : 5 : 6 = 650,000 : 1 : 6500 : 5 b-Ketoacid and malonic acid undergoes facile decarboxylation. Therefore, ethyl acetoacetate and diethyl malonate are synthetic equivalents of acetone and acetic acid. Dilithium derivatives of acetoacetic acid is also a synthetic equivalent of acetone enolate. Hydrolysis step is unnecessary, and decarboxylation can be done directly. Alkylation also can be carried out using silyl enol ethers by reaction with fluoride ion such as tetraalkylammonium fluoride salts. Little steric difference between two faces, upper and lower faces. trans cis 1/1 ratio of the prducts Pseudoaxial conformation because of allylic strain The upper face of the enolate presents three hydrogens in a 1,3-diaxial relationship to the approaching electrophile. (lower face are equatorial) Axial attack from the lower face leads directly to the chair conformation of the product. 1,3-diaxial interation with the approaching electrophile. A strong preference for alkylation to give the cis ring junction According to molecular mechanicsm the minimum-energy conformation of the enolate is a twist-boat conformation Intramolecular ring-closure reaction J is more favorable than K due to the ring strain 1.5 Generation and Alkylation of Dianions Second deprotonation Ref. Scheme 1.8 First deprotonation 1.6 Medium Effect in the Alkylation of Enolates DMF and DMSO are effective in enhancing the reactivity of enolate anions, polar aprotic solvent. Polar aprotic solvents possess excellent metal-cation coordination ability,so they can solvate and dissociates and other carbaions from ion pairs and clusters. Polar aprotic solvents are good cation solvators and poor anion solvators. Polar protic solvents coordinate to both the metal cation and the enolate ion. Water, alcohol, or ammonia Polar protic solvents are less reactive than the same enolate in a polar aprotic solvent such as DMSO. Despite the somewhat reduced reactivity of aggregated enolates, THF and DMF are the most commonly used solvents for the synthetic reactions involving (kinetic) enolate alkylation. Enolate can be enhanced by adding a reagent that can bind a alkali-metal Cations: HMPA, tetramethylenediamine(TMEDA), crown ethers. 12-crown-4; Li, 18-crown-6; Na, K. Mg2+ < Li+ < Na+ < K+ : reactivity order of enolate; the smaller, the harder strongly bind to oxygen 1.7 Oxygen versus Carbon as the Site of Alkylation Enolate anions are ambient nucleophile. O-alkylation, when the enolate is dissociated. Leaving-group effects on the C- or O-alkylation: hard-soft-acid-base(HSAB) Oxygen is harder than carbon. Oxygen leaving group such as sulfonate and sulfate are harder: reacts at the hard oxygen site of the enolate. The amount of O-alkylation is amximized by use of an alkyl sulfate or alkyl sulfonate in a polar aprotic solvent. And that of C-alkylation is maximized by an alkyl halide in a less polar solvent such as THF or DME. With 5-membered rings, colinearity cannot be achieved easily. The transition state for O-alkylation involves an oxygen lone-pair orbital and is less strained than the transition state for C-alkylation. The kinetically preffered site for both protonation and alkylation is the a-carbon The a carbon has a great negative charge compared with g carbon. Strong preference for O-alkylation in Phenoxide ions because C-alkylation disrupts aromatic conjugation Phenoxides undergo O-alkylation in solvent such as DMSO, DMF, ethers, and alcohols. However, in water and trifluoroethanol, extensive C-alkylation occurs. 1.8 Alkylation of Aldehyde , Esters, Amides, and Nitriles Alkylation of aldehyde enolate is not very common because of facile adol condensation by base. But rapid, quantitative formation of enolate avoids this: KNH2 in NH3, KH in THF. Alkylation of simple esters requires a strong base: weak base such as alkoxide promotes condensation reaction. Strong base: LDA, hesamethyldisilylamide (KHMDS). Ref. Scheme 1.9 Enolate of N-acyl oxazolidinones Diastereomeric Mixture: 95/5 Easily separated Further hydrolysis or alcoholysis Further hydrolysis or alcoholysis Final product would be 99% enantiomeric purity. Good chiral auxiliary Analgestic substance 1.9 The Nitrogen Analogs of Enols and Enolates-Enamines and Imine Anions Imine is the nitrogen analog of ketone and aldehyde. Removed by azeotropic distillation For secondary amine, vinylamine or enamine is formed. Strong dehydrating reagents to drive the reaction to completion: TiCl4 or Triethoxysilane. N-Timethylsilyl derivative: strong affinity of silicone for oxygen than nitrogen. The b-carbon atom of an enamine is a nucleophilic site because of conjugation With the nitrogen atom. Alkylation of enamine Pyrrolidine enamine Preferred enamine A serious nonbonded repulsion (A1,3 strain) destabilizes isomer 7. Because of the predominance of the less substituted enamine, alkylation occur primarily at the less substituted a carbon. trans Imine can be deprotonated at the carbon by strong base to give the nitrogen analog of enolates: imine anions or metalloenamines. Isoelectronic and structurally analogous to both enolaes and allyl anions and can also be called azaallyl anions. In toluene it exists as dimeric form, but at high THF concentration, the monomer Is favored. Just as enamines are more nucleophilic than enols, imine anions are more nucleophilic than enolates and react efficiently with alkyl halides. The nitrogen substituent R’ is syn to the double bond are the more stable. more stable Lithiated ketimines room temperature: thermodynamic composition is established. less substituted isomer: the most stable structure. Table 1.3 entry 2 : a) chelation of the methoxy group with the lithium ion b) The interaction of the lithium with the bromide c) the steric effect of the benzyl group O + OMe OMe N N N NH2 proline LDA Hydrazone is hard to be hydrolyzed. OMe N N I Li N N Li H 1) CH3I 2) H3O+ O H OMe OMe Hydrazones are more stable than alkylimines. Kinetically deprotonated Enantioselective synthesis of carboxylic acid 1.10 alkylation of Carbon Nucleophile by Conjugate Addition (Michael reaction) A catalytic amount of base is use: thermodynamic control of enolate formation W Electrophile W Nucleophile: amine, alkoxide, sulfide anions Common nucleophile: b-ketoester, maolonate esters. Fluoride ion is an effective catalyst for Michael additions involving acidic carbon compounds: excess use of fluoride because of formation of [F-H-F-] O CO2CH3 HO OLi + O + OCH3 CO2CH3 -78oC 88% 5% 25oC 7% 85% Hindered Aluminum tris(2,6-diphenylphenoxide) is an effective promoter. Ketone enolates react with enone to give 1,5-diketones.ref scheme 1.12 Quaternary carbon atom centers are easily generated. (kinetically controlled enolate) Z-enolate favors anti-adduct and E-enolate favors syn-adduct. Chelated transition state The stereoselectivity can be enhanced by addition of Ti(O-i-Pr)4. Much larger Ti(O-i-Pr)4 group replaces Li+. When the conjugate addition is carried out under kinetic conditions with stoichiometric formation of the enolate, the adduct is also an enolate until the reaction is quenched with a proton source. Tandem reaction is possible. Tandem conjugate addition reaction is an efficient means of introducing both a and b substituents at enones. Trimethylsilyl enol ether can be used with TiCl4. -78oC CH3 N O O O Ti "aci" tautomer The initial adduct is trapped in cyclic form by trimethylsilylation. Other Lewis acid Lanthanide salts catalyze addition of a-nitroesters even in aqueous solution. Alcoholic solution of potassium or sodium cyanide Triethylaluminum-hydrogen cyanide and diethylaluminum cyanide More reactive Aluminum reagent might act as a Lewis acid at the carbonyl center With chiral oxazoline, 30-50% diastereomeric excess (d.e.) can be achieved. The addition of enamines of cyclohexanones show a strong preference for attack from the axial direction, because the pi-orbital of the enamine is the site of nucleophilicity.