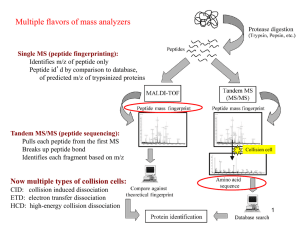
Protein Sequence Databases,
Peptides to Proteins, and
Statistical Significance
Nathan Edwards
Department of Biochemistry and Mol. & Cell. Biology
Georgetown University Medical Center
Protein Sequence Databases
• Link between mass spectra and proteins
• A protein’s amino-acid sequence provides
a basis for interpreting
•
•
•
•
Enzymatic digestion
Separation protocols
Fragmentation
Peptide ion masses
• We must interpret database information as
carefully as mass spectra.
2
More than sequence…
Protein sequence databases provide much
more than sequence:
•
•
•
•
•
Names
Descriptions
Facts
Predictions
Links to other information sources
Protein databases provide a link to the current
state of our understanding about a protein.
3
Much more than sequence
Names
• Accession, Name, Description
Biological Source
• Organism, Source, Taxonomy
Literature
Function
• Biological process, molecular function,
cellular component
• Known and predicted
Features
• Polymorphism, Isoforms, PTMs, Domains
Derived Data
• Molecular weight, pI
4
Database types
Curated
• Swiss-Prot
• UniProt
• RefSeq NP
Translated
• TrEMBL
• RefSeq XP, ZP
Omnibus
• NCBI’s nr
• MSDB
• IPI
Other
• PDB
• HPRD
• EST
• Genomic
5
SwissProt
• From ExPASy
• Expert Protein Analysis System
• Swiss Institute of Bioinformatics
• ~ 515,000 protein sequence “entries”
• ~ 12,000 species represented
• ~ 20,000 Human proteins
• Highly curated
• Minimal redundancy
• Part of UniProt Consortium
6
TrEMBL
• Translated EMBL nucleotide sequences
• European Molecular Biology Laboratory
• European Bioinformatics Institute (EBI)
• Computer annotated
• Only sequences absent from SwissProt
• ~ 10.5 M protein sequence “entries”
• ~ 230,000 species
• ~ 75,000 Human proteins
• Part of UniProt Consortium
7
UniProt
• Universal Protein Resource
• Combination of sequences from
• Swiss-Prot
• TrEMBL
• Mixture of highly curated/reviewed
(SwissProt) and computer annotation
(TrEMBL)
• “Similar sequence” clusters are available
• 50%, 90%, 100% sequence similarity
8
RefSeq
• Reference Sequence
• From NCBI (National Center for
Biotechnology Information), NLM, NIH
• Integrated genomic, transcript, and
protein sequences.
• Varying levels of curation
• Reviewed, Validated, …, Predicted, …
• ~ 9.7 M protein sequence “entries”
• ~ 209,000 reviewed, ~ 90,000 validated
• ~ 39,000 Human proteins
9
RefSeq
• Particular focus on major research
organisms
• Tightly integrated with genome projects.
• Curated entries: NP accessions
• Predicted entries: XP accessions
• Others: YP, ZP, AP
10
IPI
• International Protein Index
• From EBI
• For a specific species, combines
• UniProt, RefSeq, Ensembl
• Species specific databases: HInv-DB, VEGA, TAIR
• ~ 87,000 (from ~ 307,000 ) human protein
sequence entries
• Human, mouse, rat, zebra fish, arabidopsis,
chicken, cow
• Slated for closure November 2010, but still
going…
11
MSDB
• From the Imperial College (London)
• Combines
• PIR, TrEMBL, GenBank, SwissProt
• Distributed with Mascot
• …so well integrated with Mascot
• ~ 3.2M protein sequence entries
• “Similar sequences” suppressed
• 100% sequence similarity
• Not updated since September 2006
(obsolete)
12
NCBI’s nr
• “non-redundant”
• Contains
•
•
•
•
•
GenBank CDS translations
RefSeq Proteins
Protein Data Bank (PDB)
SwissProt, TrEMBL, PIR
Others
• “Similar sequences” suppressed
• 100% sequence similarity
• ~ 10.5 M protein sequence “entries”
13
Human Sequences
• Number of Human
genes is believed to
be between 20,000
and 25,000
SwissProt
~ 20,000
RefSeq
~ 39,000
TrEMBL
~ 75,000
IPI-HUMAN ~ 87,000
MSDB
~130,000
nr
~230,000
14
DNA to Protein Sequence
Derived from http://online.itp.ucsb.edu/online/infobio01/burge
15
UCSC Genome Browser
• Shows many
sources of protein
sequence
evidence in a
unified display
16
Accessions
•
•
•
•
•
Permanent labels
Short, machine readable
Enable precise communication
Typos render them unusable!
Each database uses a different format
•
•
•
•
Swiss-Prot: P17947
Ensembl: ENSG00000066336
PIR: S60367; S60367
GO: GO:0003700;
17
Names / IDs
•
•
•
•
Compact mnemonic labels
Not guaranteed permanent
Require careful curation
Conceptual objects
• ALBU_HUMAN
• Serum Albumin
• RT30_HUMAN
• Mitochondrial 28S ribosomal protein S30
• CP3A7_HUMAN
• Cytochrome P450 3A7
18
Description / Name
• Free text description
• Human readable
• Space limited
• Hard for computers to interpret!
• No standard nomenclature or format
• Often abused….
• COX7R_HUMAN
• Cytochrome c oxidase subunit VIIarelated protein, mitochondrial [Precursor]
19
FASTA Format
•>
• Accession number
• No uniform format
• Multiple accessions separated by |
• One line of description
• Usually pretty cryptic
• Organism of sequence?
• No uniform format
• Official latin name not necessarily used
• Amino-acid sequence in single-letter code
• Usually spread over multiple lines.
20
FASTA Format
21
Organism / Species /
Taxonomy
• The protein’s organism…
• …or the source of the biological sample
• The most reliable sequence annotation
available
• Useful only to the extent that it is correct
• NCBI’s taxonomy is widely used
• Provides a standard of sorts; Heirachical
• Other databases don’t necessarily keep up
• Organism specific sequence databases
starting to become available.
22
Organism / Species /
Taxonomy
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
•
Buffalo rat
Gunn rats
Norway rat
Rattus PC12 clone IS
Rattus norvegicus
Rattus norvegicus8
Rattus norwegicus
Rattus rattiscus
• Rattus sp.
23
Rattus sp. strain Wistar
Sprague-Dawley rat
Wistar rats
brown rat
laboratory rat
rat
rats
zitter rats
Controlled Vocabulary
• Middle ground between computers and
people
• Provides precision for concepts
• Searching, sorting, browsing
• Concept relationships
• Vocabulary / Ontology must be established
• Human curation
• Link between concept and object:
• Manually curated
• Automatic / Predicted
24
Gene Ontology
• Hierarchical
• Molecular function
• Biological process
• Cellular component
• Describes the vocabulary only!
• Protein families provide GO association
• Not necessarily any appropriate GO category.
• Not necessarily in all three hierarchies.
• Sometimes general categories are used because
none of the specific categories are correct.
25
Gene Ontology
26
Protein Families
• Similar sequence implies similar function
• Similar structure implies similar function
• Common domains imply similar function
• Bootstrap up from small sets of
proteins/domains with well understood
characteristics
• Usually a hybrid manual / automatic
approach
27
Protein Families
28
Protein Families
29
Sequence Variants
• Protein sequence can vary due to
• Polymorphism
• Alternative splicing
• Post-translational modification
• Sequence databases typically do not
capture all versions of a protein’s
sequence
30
Swiss-Prot
Variant Annotations
31
Swiss-Prot
Variant Annotations
32
Omnibus Database
Redundancy Elimination
• Source databases often contain the same
sequences with different descriptions
• Omnibus databases keep one copy of the
sequence, and
• An arbitrary description, or
• All descriptions, or
• Particular description, based on source preference
• Good definitions can be lost, including
taxonomy
33
Description Elimination
• gi|12053249|emb|CAB66806.1|
hypothetical protein [Homo sapiens]
• gi|46255828|gb|AAH68998.1|
COMMD4 protein [Homo sapiens]
• gi|42632621|gb|AAS22242.1|
COMMD4 [Homo sapiens]
• gi|21361661|ref|NP_060298.2|
COMM domain containing 4 [Homo sapiens]
• gi|51316094|sp|Q9H0A8|
COM4_HUMAN COMM domain containing protein 4
• gi|49065330|emb|CAG38483.1|
COMMD4 [Homo sapiens]
34
Peptides to Proteins
Nesvizhskii et al., Anal. Chem. 2003
35
Peptides to Proteins
36
Peptides to Proteins
• A peptide sequence may occur in
many different protein sequences
• Variants, paralogues, protein families
• Separation, digestion and ionization is
not well understood
• Proteins in sequence database are
extremely non-random, and very
dependent
37
Indistinguishable Protein
Sequences
38
Nesvizhskii, Aebersold, Mol Cell Proteomics, 2005
Indistinguishable Protein
Sequences
39
Nesvizhskii, Aebersold, Mol Cell Proteomics, 2005
Protein Families
40
Nesvizhskii, Aebersold, Mol Cell Proteomics, 2005
Protein Grouping
Scenarios
• Parsimony
• Minimum # of proteins
• Weighted
• Choose proteins
with the most confident
peptides
(ProteinProphet)
• Show all
• Mark repeated peptides
• Often no (ideal)
resolution is possible!
Nesvizhskii, Aebersold, Mol Cell Proteomics, 2005
41
High Quality Peptide
Identification: E-value < 10-8
42
Moderate quality peptide
identification: E-value < 10-3
43
Peptide Identification
• Peptide fragmentation by CID is poorly
understood
• MS/MS spectra represent incomplete
information about amino-acid sequence
• I/L, K/Q, GG/N, …
• Correct identifications don’t come with a
certificate!
44
Peptide Identification
• High-throughput workflows demand we
analyze all spectra, all the time.
• Spectra may not contain enough
information to be interpreted correctly
• …bad static on a cell phone
• Peptides may not match our assumptions
• …its all Greek to me
• “Don’t know” is an acceptable answer!
45
What scores do “wrong”
peptides get?
• Generate random peptide sequences
• Real looking fragment masses
• Empirical distribution
• Require similar precursor mass
• Arbitrary score function can model
anything we like!
46
Random Peptide Scores
Fenyo & Beavis, Anal. Chem., 2003
47
Random Peptide Scores
Fenyo & Beavis, Anal. Chem., 2003
48
Random Peptide Scores
• Truly random peptides don’t look much like
real peptides
• Just use peptides from the sequence
database!
• Assumptions:
• IID sampling of “score” values per spectra
• Caveats:
• Correct peptide (non-random) may be included
• Peptides are not independent
49
Extrapolating from the
Empirical Distribution
• Often, the empirical shape is
consistent with a theoretical model
Fenyo & Beavis, Anal. Chem., 2003
Geer et al., J. Proteome Research, 2004
50
E-values vs p-values
• Need to adjust for the size of the sequence
database
• Best false/random score goes up with number
of trials
• E-value makes this adjustment
• Expected number of incorrect peptides (with
this score) from this sequence database.
• E-value = # Trials * p-value (to 1st approx.)
51
False Discovery Rate
• Which peptide IDs to accept?
• E-value only provides a per-spectrum statistic
• With enough spectra, even these can be
misleading!
• Decide which spectra (w/ scores) will be
accepted:
• SEQUEST Xcorr, E-value, Score, etc., plus...
• Threshold on identification criteria
• Control the proportion of incorrect
identifications in the result for entire dataset
52
Distribution of scores
over all spectra
200
180
160
140
120
100
80
60
40
20
0
-3.9
-2.3
-0.7
0.9
2.5
53
4.1
5.7
7.3
Brian Searle, Proteome Software
Distribution of scores
over all spectra
200
False
180
160
140
120
100
80
True
60
40
20
0
-3.9
-2.3
-0.7
0.9
2.5
54
4.1
5.7
7.3
Brian Searle, Proteome Software
False Discovery Rate
• FDRscore ≥ x = # false ids with score ≥ x
# all ids with score ≥ x
• Need to estimate numerator!
• Assumes the false (and true) scores,
sampled over spectra, are IID
• Not true for some peptide-spectrum scores
• (Mostly) true for E-values
• Can compute the # false ids using a decoy
search…
55
Peptide Prophet
Keller et al., Anal. Chem. 2002
Distribution of spectral scores in the results
56
Decoy searches
• Shuffle or reverse sequence database
• Same size as original
• Known false identifications
• Estimate “False” distribution
• Alternatively, merge target+decoy results:
• Competition between target and decoy scores
• Assume false target and false decoys each
win half the time
• FDRscore ≥ x = 2 * # decoy ids with score ≥ x
# target ids with score ≥ x
57
Summary
• Protein sequence databases have
varying characteristics, choose wisely!
• Inferring proteins from peptides can be
(very) tricky!
• Statistical significance can help control
the proportion of errors in the (peptidelevel) results.
58