Experiment IV Mass Spectrometry of Proteins and Peptides This is a

EXPERIMENT IV
MASS SPECTROMETRY OF PROTEINS AND PEPTIDES
This is a demonstration exercise, followed by a good bit of data analysis work. The demonstrators will show you how to acquire MALDI, Electrospray (ESI), and MS/MS spectra (both CAD and ETD (see below)) of proteins and the tryptic digest mixtures of peptides from those proteins. In order to be allowed to use these instruments on your own, you will need to be trained by Dr. Lijiang Song, which can occur if/when you join a group for PhD study.
The proteins involved will come from this list: Lysozyme, Ribonuclease A, α-lactoctoglobulin, bovine serum albumin, myoglobin, α-chymotrypsin, calmodulin, and cytochrome-c. The source is usually bovine. Be sure to look up the sequences of these proteins and be ready to calculate their masses, the masses of all tryptic peptides from these proteins, and the ABC-XYZ series of masses from these peptides (this will be described in the lectures). Comparison of these calculated masses to the measured ones is the key to this experiment.
Tasks:
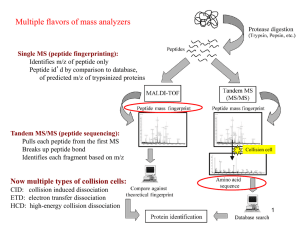
1. The demonstrators will generate MALDI and ESI mass spectra of whole proteins and provide them to you. You will need to load these spectra onto your computer using a plotting program such as
Matlab or Origin, and analyze the spectra yourself. Calculate and report the masses of these proteins. Which proteins are they?
2. The demonstrators will generate MALDI and ESI mass spectra of the tryptic digest peptide mixtures, which is a standard experiment called a "peptide mass fingerprint (PMF)". As above, load the spectra and analyze the spectra. Determine the masses of all the peptides, and compare to the calculated masses of tryptic peptides from the protein list above. Which proteins are they?
3. Several the tryptic peptides from task 2 will be fragmented using Collisionally Activated
Dissociation (CAD) and Electron Transfer Dissociation (ETD). This is a standard experiment known as
Tandem Mass Spectrometry or MS/MS (or MS n sometimes). As above, load the spectra and analyze the spectra. Determine the masses of all significant peaks in each spectrum. Determine the sequence of each peptide (note: the CAD and ETD data are often complementary and can be used to double check your assigned sequence), and report it.
Questions:
1. Is analysis of the whole protein mass useful?
2. How would the whole protein mass, PMF, and MS/MS experiments change if the protein was post-translationally modified (PTM'd), for example truncated (loss of a signal peptide), phosphorylated, acetylated, deamidated?
3. Are the fragment ion products from CAD or ETD even-electron, or odd-electron molecular species? How can you tell the difference?
4. What is a complementary pair? A golden complementary pair? How can they be used?
5. Which masses in the MS/MS spectra are the most useful in your analysis?
6. In this case, we have a very limited number of possible proteins in our list, but humans can make an estimated 30,000 proteins, which expands to several hundred thousand if PTMs are counted separately. Would this analysis work in such a big search space?
7. In this case, we only do MS/MS. How would MS/MS/MS or higher orders of MS n be useful?
Which kinds of instruments are capable of such experiments?
Report:
The report should follow the usual format for a scientific manuscript:
I. Introduction
What is the goal of this experiment?
Important background and theory
Describe the instruments involved
How would you use this kind of experiment in a chemistry, biochemistry, or medical research environment?
II. Methods a. What instruments were used, what type were they, what models? b. What solution conditions for ESI were used, flow rates, MS/MS parameters? c. What matrix was used? What was the approximate mole ratio with the analyte protein/peptide? What laser power?
III. Experimental results (all data and spectra)
For each spectrum (there should be at least 5-6 such spectra) that you analyze, make a figure from the spectrum in powerpoint and label it with all the appropriate information. This should definitely include masses and assignments. Use an Excel spreadsheet to make a table of all observed peak masses for each spectrum, use it to assign the identity of the peaks, calculate the mass of each assigned structure, and calculate the mass error in ppms. This table is a powerful tool for checking the accuracy of your assignment.
The report should include all figures and tables.
Report the protein identities from above.
IV. Discussion
For the PMF experiments, create a figure of the assigned protein sequence, underlining or highlighting the peptides that could be assigned. For the MS/MS experiments, create a figure of the peptide sequence, showing the assigned cleavages (ABC/XYZ).
Discuss the meaning of these plots, answer the questions from above.
V. Conclusion
Did it work?
What was learned?
How can this be used in your research?