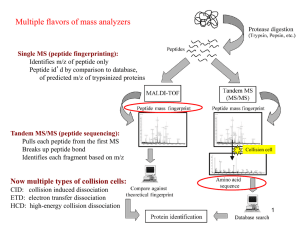
Changes in Protein Abundance Across Nutrient
advertisement

Changes in Protein Abundance Across Nutrient Conditions Sean Hackett, Jonathan Goya, David Perlman and Josh Rabinowitz Background Across varying nutrient conditions, with differing limiting nutrients and maximal growth rates, cells must deal with two challenges: ① Increasing fluxes (rates of enzymatic reactions) in parallel with nutrient availability ② Sensing which nutrients are limiting and growing optimally with respect to that limitation To study this control of metabolism, previous researchers in the Botstein and Rabinowitz labs have measured transcript and metabolite abundance across 25-36 steady state conditions. • Limiting nutrients: glucose (C), ammonium (N), phosphate (P), uracil (U) and leucine (L) (in auxotrophs). • Dilution rates: 0.05, 0.11, 0.16, 0.22, 0.30 h-1 Transcriptomics Going from peptides to proteins Working with peptides Of the 11434 peptides that were measured in at least one of the 15 conditions, the coverage of peptides that are quantifiable under all 15 conditions is quite poor We need methods that are: • Able to use peptides with missing data to understand patterns in complete data • Look at patterns of missing data to identify peptides that are present in only a subset of conditions Number of conditions where a peptide is quantifiable Overlap of peptides measured in both conditions relative to just one • Remove all peptides with more than 0.2 fraction of missing data • Remove all peptides with less than 32768 ions measured • This results in 5106 peptides that are retained and 6328 peptides which are discarded. When using relative variation in peptides to predict variation in protein abundance we need to deal with two factors ① If a peptide maps to multiple proteins, it should be attributed to a protein (thereby adding signal) to the extent that its pattern matches the pattern from other peptides. ② Some peptides won't conform to the trends of their protein because they may be the noncovalently modified complement of a set of unascertained modified peptides. These peptides shouldn't inform the general protein trend, and may be interesting to analyze in isolation. Metabolomics Examples of fits Comparing transcripts and proteins Raw Ω Impute missing values with PCA-based method Brauer 2008 Boer 2010 In a followup to these studies, we are combining these metabolic and transcriptional datasets with quantitative proteomics, rates of nutrient uptake and secretion and composition measurements. Experimental protocol: - Grow FY4 S. cerevisiae to steady state under the desired nutrient limitation and dilution rate - Filter culture and freeze on li-N2 - Homogenize and extract proteins - Combine sample proteins 1-1 with 15N labeled reference chemostat (p0.05) - Digest combined proteins with trypsin - Fractionate peptides into 24 fractions using isoelectric focusing - Identify and quantify peptides using time of flight tandem mass spectrometry on an Agilent Q-TOF Computational (by JG): - MS1 data (peaks in 2D: elution time * mass-to-charge) gives total ions, with uncertainty about the identity of the ion - Compare MS2 data (peptide sequences) to predicted peptides based on protein sequences, passing identity to some MS1 peaks - Align MS1 data across files to pass identities between peaks identified in only a subset of conditions From brauer 2008 infer relative abundances at dilution rates attained in this study How correlated are proteins and their corresponding transcripts ? Future Directions • What types of proteins are present or absence at high and low growth rates, or are limitation-specific • What types show a quantitative relationship to growth rate or a limitation-specific effect • What is responsible for the poor correlation of many proteins vs. transcripts • How much can these patterns be explained by motifs in DNA, RNA or proteins • Modeling of fluxes and studying metabolic versus hierarchical control and the sufficiency of established reaction mechanisms