Informatics for proteomic
inventories
david.l.tabb@vanderbilt.edu
Biomedical Informatics
Vanderbilt University
Overview
• Explaining the whys and hows of proteomics
• Matching peptides from protein sequence
databases to MS/MS spectra
• Filtering peptide-spectrum matches (PSMs) to
an acceptable false discovery rate (FDR)
• Inferring proteins parsimoniously and scalably
Methods capture only part of story
Genomics and epigenetics
describe state of “catalog.”
Proteomics measures current
inventory of cell capabilities.
Transcriptomics describes
current “purchase orders.”
Metabolomics examine
cell state most directly.
J_Alves: glycine tRNA
J_Alves: glucose and cholesterol
ElaineMeng: H-ras, PDB 121P
What does proteomics include?
1D and 2D Gel
Electrophoresis
Protein
Inventories
Protein
Quantitation
Tissue
Imaging
Post-Translational
Modifications
Gerald_G scales, Gsagri04: gel,
AB SCIEX tissue image
Discovery Proteomics
Protein
Mixture
High-Resolution
Mass Spectrometry
Peptide
Mixture
Isolate
Ions of Peptide
Peptide
Fractionation
Liquid
Chromatography
Collide Ions to
Dissociate
Electrospray
Ionization
Collect Fragments
in Tandem MS
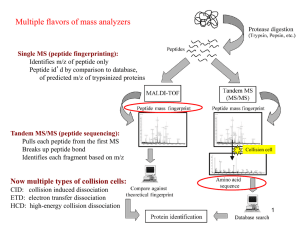
Two types of measurements for each peptide: intact m/z
(mass/charge) and a list of fragment m/zs.
Collision-induced dissociation (CID)
O
+
R1
OH
R3
O
NH
R3
O
NH
H2N
NH2
R2
OH
+
O
R2
O
R4
N
H
R1
O
H2N
NH
+
O
OH
R4
NH2
• “Tickle” energizes peptide, causing varied
conformations and proton movement.
• A mobile proton associates with a carbonyl
adjoining a peptide bond, drawing electrons.
• Electrons of the prior carbonyl attack, forming
a ringed intermediate that quickly dissociates.
Wysocki et al, Anal. Chem. (2000) 35: 1399-406.
Paizs and Suhai, Rapid Comm. Mass Spectrom. (2002) 16: 1699-1702.
Broken peptide bonds yield fragments
TSIIGTIGPK
N-terminal
b4 ion
C-terminal
y6 ion
HFISELEK, +2 charge state
Neutral loss of
water from peptide
-ISELEK
-LEK
HF-
-SELEK
-FISELEK
Same spectrum compared to
FHEIKELS instead of HFISELEK
Neutral loss of
water from peptide
FH- has same
mass as HF-
-EIKELS has same
mass as -ISELEK
Disassembly and reassembly
Mixture of Proteins
Confidently identified
proteins
...YGR192C
YGR204W
YGR208W
YGR209C...
Mixture of Peptides
Confidently identified
peptide sequences
...LSEGTSFR
LSELIGAR
LSENLRK
LSEPVHK...
Collection of tandem
mass spectra
Collection of raw
peptide identifications
LSELIGAR
z=2 XCorr=3.5
After AI Nesvizhskii, Mol Cell Proteomics (2005) 4: 1419-40.
Database search overview
Eng et al (1994) J. Amer. Soc. Mass Spectrom. 5: 976-989.
Yates et al (1995) Anal. Chem. 67: 1426-1436.
Emulating proteases in silico
N Edwards and R Lippert. Lecture Notes In Computer Science (2002) 2452: 68-81.
Dynamic PTMs grow search space
Because multiple
PTMs may be in
each peptide,
adding PTMs to a
search creates an
exponential cost.
Here, three sites
lead to eight PTM
variants.
CASA1_BOVIN
Peptide mass filter
• Sequences outside mass
tolerance are not compared.
• Many sequences may share
a common mass.
• Sequences of one mass may
score differently.
• Sequences of different mass
may score the same.
Sequence m/z delta (ppm) Fragment Score
KDTLTSR
-15.69860403
N/A
DKTLTSR
-15.69860403
N/A
KLCIM*R
-14.64112528
N/A
KLCLM*R
-14.64112528
N/A
RDRFAR
-14.07051821
N/A
RAFRDR
-14.07051821
N/A
RVM*RSR
-9.966599813
37.70
RVRM*SR
-9.966599813
37.70
RSTITSR
-2.023663496
72.39
TSRLTSR
-2.023663496
48.14
RITSSTR
-2.023663496
36.39
RLTSSTR
-2.023663496
36.39
RTLTSSR
-2.023663496
36.39
SITRTSR
-2.023663496
35.57
RTSSTIR
-2.023663496
35.24
RTSSTLR
-2.023663496
35.24
RSSTLTR
-2.023663496
31.32
HHKRSR
-0.395577679
30.18
LFQAVSR
2.873416767
34.95
APPPVPSR
2.873416767
34.39
PKYLGSR
2.873416767
29.64
KIM*LGSR
6.977335166
34.95
LM*KIGSR
6.977335166
29.16
KLIGM*SR
6.977335166
28.00
Fragment masses and
charge segregation
H+
+2
AA
H+
AA
AA
H
H+
AA
+3
AA
H+
AA
H+
AA
AA
AA
H
AA
AA
OH
H+
AA
H+
AA
AA
AA
OH
H+
H
AA
AA
AA
OH
Sequest cross correlation
• Normalize observed spectrum.
• Generate model spectrum for each candidate.
• Convert observed and model spectrum to
frequency domain by FFT.
• Cross-correlate, reporting ratio between zerooffset alignment and nearby alignments.
J Eng et al. J. Proteome Res. (2008) 7: 4598-4602.
J Eng et al. J Amer. Soc. Mass. Spectrom. (1994) 5: 976-989.
X!Tandem scoring
•
•
•
•
•
•
Predict more accurate fragment intensities
Count matched b ions and matched y ions
Compute dot product of intensities
Generate hyperscore = B!Y!
ObsExp
Build histogram of scores per spectrum
Report expectation value
Craig and Beavis. Rapid Comm. Mass Spectrom. (2003) 17:2310-2316.
Fenyö and Beavis. Anal. Chem. (2003) 75: 768-774.
Random match probabilities
• Imagine spectrum as jar of 100 black and 900
white marbles (peaks and voids).
• Sample 20 marbles for a predicted peaklist,
drawing 15 black and 5 white.
• Compute probability of random match by
hypergeometric distribution:
100 900
15 5
p
3.63146E- 12
1000
20
T Fridman. J. Bioinfo. Computat. Bio. (2005) 3: 455-476.
Disassembly and reassembly
Mixture of Proteins
Confidently identified
proteins
...YGR192C
YGR204W
YGR208W
YGR209C...
Mixture of Peptides
Confidently identified
peptide sequences
...LSEGTSFR
LSELIGAR
LSENLRK
LSEPVHK...
Collection of tandem
mass spectra
Collection of raw
peptide identifications
LSELIGAR
z=2 XCorr=3.5
After AI Nesvizhskii, Mol Cell Proteomics (2005) 4: 1419-40.
The “longest list” problem
• Perceived value of early proteomics
experiments was linked only to sensitivity.
• Systems to evaluate specificity lagged behind,
and false positive rates were left unchecked.
• Two developments were needed:
– Community consensus on reporting standards
– New tools for evaluating identification error rates
Carr et al. Mol. Cell. Proteomics (2004) 3: 531-533.
Taylor et al. Nature Biotech. (2007) 25: 887-893
Strategy I: Target/decoy estimates FDR
• Sequence database has equal numbers of
target and decoy sequences.
• False IDs distribute evenly between target and
decoy sequences.
• Apply a threshold, and:
– False estimate = 2 x [decoy hit count].
– False Discovery Rate (FDR) = False estimate
divided by number of passing IDs.
Elias and Gygi.
Nature Methods
(2007) 4: 207-214
Decoys model false distribution
• A match to targets is
possibly true; a match to
decoys is surely false.
• As threshold slides to
lower scores, more decoys
are kept, escalating FDR.
• Alternatively,
𝑑𝑒𝑐𝑜𝑦
𝑡𝑎𝑟𝑔𝑒𝑡
may
be used if decoys are
excluded from final list.
Elias Nat. Methods (2007) 4: 207-214
Strategy II: Peptide Prophet
• Estimates correctness probability for
individual identifications
• Combines multiple subscores from each
Sequest identification through DFA
• Fits mixed model to observed matches with
expectation maximization
• A Keller. Anal. Chem. (2002) 74: 5383-5392.
Discriminant Function Analysis
combines sub-scores from Sequest
Mixture Model analysis
separates true and false distributions
• Expectation
maximization
adjusts two curves
to fit observed data.
• Here, negatives are
fit to a gamma
distribution and
positives to a
normal distribution.
Disassembly and reassembly
Mixture of Proteins
Confidently identified
proteins
...YGR192C
YGR204W
YGR208W
YGR209C...
Mixture of Peptides
Confidently identified
peptide sequences
...LSEGTSFR
LSELIGAR
LSENLRK
LSEPVHK...
Collection of tandem
mass spectra
Collection of raw
peptide identifications
LSELIGAR
z=2 XCorr=3.5
After AI Nesvizhskii, Mol Cell Proteomics (2005) 4: 1419-40.
Why are peptides shared
among proteins?
“Orthologs are direct evolutionary counterparts
derived from a common ancestor through
vertical descent; whenever we speak of the ‘the
same gene in different species,’ we actually
mean orthologs. In contrast, paralogs are genes
within the same genome that have evolved by
duplication.”
Koonin. Genome Biology (2001) 2: comment 1005.1-1005.2.
Protein isoforms
• A single gene may give rise to many transcripts
that overlap for one or more exons.
• When isoforms are listed as separate proteins
in the FASTA, a peptide may match a shared or
distinctive part of a protein sequence.
• VEGF incorporates eight exons, where either 6
or 7, both, or neither may be incorporated.
Parsimony
• noun: “economy of explanation in conformity
with Occam's razor”
– Merriam Webster OnLine
• “Plurality ought never be posed without
necessity.”
– William of Occam
IDPicker
1. Assemble maximal protein list.
2. Combine proteins that point to the same
peptides, and combine peptides that point to
the same proteins.
3. Find “set cover” by greedy algorithm to pick
minimal protein list to explain peptides.
B Zhang et al. J. Proteome Res. (2007) 6: 3549-3557.
Z Ma et al. J. Proteome Res. (2010) 8: 3872-3881.
Two proteins or seven?
• Sample mixes mouse
and human proteins.
• Isoforms, paralogs, and
orthologs complicate
protein-peptide map.
• Untangling relationships
is non-trivial.
Data from Broad Institute, CPTAC
Greedy algorithm
Data from Broad Institute, CPTAC
ProteinProphet
1. Combine peptide identification probabilities
into protein identification probabilities.
2. Distribute probability for shared peptides
across multiple proteins.
3. Compute protein probability by subtracting
probability that all observed peptides are
false from 1.
– AI Nesvizhskii. Anal. Chem. (2003) 75: 4646-4658.
Number of Sibling Peptides and
Degenerate Peptides
• NSP places more confidence in peptides for
proteins with abundant supporting evidence.
• Degenerate peptides match multiple potential
proteins, each associated with a weight.
• Expectation maximization determines weights
that minimize proteins count and maximize
protein probability.
Parsimony reduces protein lists
Maximal list
Grouping indiscernibles
Grouping + parsimony
SwissProt HUMAN
International Protein Index
SwissProt Multispecies
Zhang et al. J. Proteome Res. (2007) 6: 3549-57.
Protein FDR is not PSM FDR
Minimum
PSMs/Prot
Confident
PSMs
Distinct
Peptides
Distinct
Empirical
Protein Groups Protein FDR
2
252251
102934
8342
0.089
3
250520
101198
7474
0.033
4
248919
99788
6923
0.014
5
247087
98253
6441
0.008
•
•
•
•
PSM FDR fixed at 3%
Two distinct peptides required per protein
True PSMs group together on true proteins.
False PSMs spread across the database.
Data from Broad Institute, CPTAC
Takeaway messages
• Tandem mass spectrometry produces lists of
fragment m/z values and precursor masses.
• Database search narrows the set of all
possible peptides to plausible candidates.
• Controlling peptide and protein FDR is
essential for credible, publishable inventories.
• Parsimony and scalable filtering are necessary
to field modern data sets.