MN-B-C 2 Analysis of High Dimensional (-omics) Data
Week 5: Proteomics 1
Kay Hofmann – Protein Evolution Group
http://www.genetik.uni-koeln.de/groups/Hofmann
Detection Proteomics
Which proteins are there? Which are abundant?
Which part of a protein is there?
Are there changes in protein presence or abundance?
Modification Proteomics
Which proteins have posttranslational modifications?
What fraction of a protein pool is modified?
Are there changes in protein modification level?
Interaction Proteomics
Which bind to each other or form complexes?
What fraction of a protein pool is bound/complexed?
Are there changes in protein interaction patterns?
Two-dimensional gel electrophoresis
Detektion (and possibly quantification) of entire proteins.
limited scope, lack of reproducibility
old school
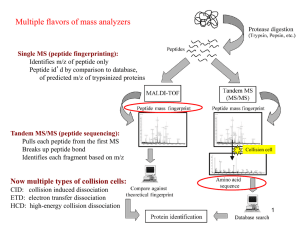
Mass spectrometry
Possibly coupled with liquid chromatography (LC)
Detection (and possibly quantification) of peptides
Requires sophisticated instrumentation
Antibodies
Candidate proteins have to be known
Specific antibodies have to be available
only suitable for small- and medium-scale studies
Specific protein tags
Candidate proteins have to be known and be modified artificially
only suitable for special applications
1st Dimension
Isoelectric focussing
Separation by
isoelectric point
2nd Dimension
SDS PAGE
Separation by size
Modification
Differences in Gel properties can be (partially) compensated
Protein Recognition
Can to (some degrees) be done directly on gel by a combination of IEP/MW-values and
recognition of spot patterns.
Usually, MS analysis of eluted spots required.
How many proteins can be detected ?
In a 4 h run on the newest generation instrument:
• 25.000 peptides
• 4.000 proteins
Sample requirement
4 µg peptide sample
Sample preparation time
6h plus digest time
x3 y3 z3
R1 O
x2 y2 z2 x1 y1 z1
R2 O
R3 O
H+
R4 O
H – N – C – C – N – C – C – N – C – C – N – C – C – OH
H H
H H
a1 b1 c 1
H
H
H H
a2 b2 c2 a3 b3 c3
a,b,c ions: charge retained in N-terminal fragment
x,y,z ions: charge retained in C-terminal fragment
The type of generated ion depends on the MS method
In principle, the fragmentation pattern of a peptide could result in a complete
series of (e.g.) b-ions that allows the determination of the peptide sequence.
Real data typically allows only identification of 3mer or 4mer peptides.
K/R
GWSV
K/R 1489.430
650.213
Short tags can identify a peptide if combined with additional data (size)
This method can tolerate some degree of modification or variability,
even if this is unknown/unexpected.
Identification of suitable tags is difficult, often done manually
The entire fragment spectrum of
a peptide is compared to a
database of expected spectra
for every possible peptide.
Possible peptides are taken
from a proteome-wide sequence
database, taking the cleaving
enyzme into account.
Even if some ions are missing or
too much, the correcpt peptide
can be identified by a good
correlation of expected and
observed pattern.
Problems with polymorphisms,
modifications, other unexpected
things.
Nowadays rarely done
Can introduce bias
Protein separation is often not very reproducible
Reduces sample/spectrum complexity
Spectra contain data of fewer proteins. But: more spectra have to be measured
Allows quantification at protein level
Protein amounts (e.g. LC peaks) more quantitative than peptide amounts or even MS
peaks.
Allows the detection of minor components
Minor proteins are not overwhelmed by peptids from major components
missed cleavage
site
matches this protein, but also
another one (with better score)
b* =
b0 =
b++ =
b without NH3
b without H2O
b with two charges
Mass Spectrometry is not a quantitative method
Different ions have different physicochemical properties, 'flyability', stability etc.
Quantification before MS
In some settings, it is possible to quantify the proteins or peptides before MS analysis,
e.g. by gels, LC.
Semi-quantitative 'label-free' approaches
While the peak intensity does not correlate with protein abundance, the peptide count
can be used for quantification (iBAQ, spectral counting, PAI)
Quantitative labeling approaches
Allow quantitative comparison of protein abundance under various conditions (iCAT,
iTRAQ, SILAC)
Peptide frequency is correlated with protein abundance
• only semi-quantitative
• requires comparison over multiple runs, conditions must be stable & reproducible
• normalization for protein size required (big proteins generate more peptides)
Observed vs. Non-observed
•Frequently used line of argumentation. Statistics difficult; effects are easily
overestimated
• Difference 6 vs 0 observations typically not significant. (Audic &Claverie statistics)
Protein abundance index (PAI)
• The number of observed peptides divided by the number of observable peptides per
protein.
• Related to the logarithm of protein abundance
Stable isotope labeling by amino acids in cell culture
• A frequently used type of metabolic labeling
One cell culture is fed with Lys/Arg
containing light C12 atoms
One cell culture is fed with Lys/Arg
containing heavy C13 atoms
Proteins from the two cultures are mixed and
analysed in a single experiment. The proteins
and resulting peptides behave identically
During MS analysis, all labeled ions appear as a duplet
with a defined size difference. The intensity ratio of
these peaks is a good proxy for the ratio of protein
abundance in the two cultures
Two commercially available variants:
TMT: Tandem Mass Tag
iTRAQ: Isobaric Tag for relative and absolute quantitation
Labeling is not done
metabolically but at the
protein or peptide level.
Isobaric properties
ensure that peptide
differences are only
observed after
fragmentation
Demonstration von
• Human Protein Atlas (http://www.proteinatlas.org)
• ArrayExpress Expression atlas (http://www.ebi.ac.uk/gxa)
 0
0