Fluorescence Microscopy and special applications.
advertisement

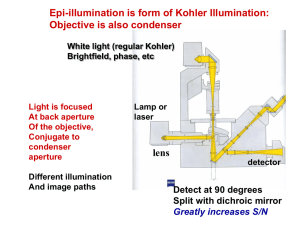
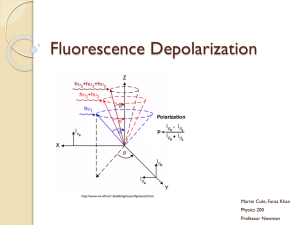
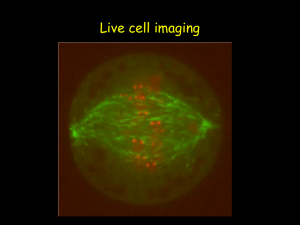
Bringing Light into the chaos: a general introduction to optics and light microscopy Part 1: The root of all evil Part 2: Contrasting techniques a reminder… • • • • • Brightfield -absorption Darkfield -scattering Phase Contrast -phase interference Polarization Contrast -polarization Differential Interference Contrast (DIC) -polarization + phase interference • Fluorescence Contrast Fluorescence techniques • Standard techniques: wide-field confocal 2-photon • Special techniques: FRET FLIM FRAP Photoactivation TIRF Fluorescence Excited state excitation emission shorter wavelength, higher energy longer wavelength, less energy Ground state Stoke’s shift Fluorophores (Fluorochromes, chromophores) • Special molecular structure • Aromatic systems (Pi-systems) and metal complexes (with transition metals) • characteristic excitation and emission spectra Excitation / emission Excitation/emission spectra always a bit overlapping filterblock has to separate them a) Exitation filter b) Dichroic mirror (beamsplitter) a) Emission filter Excitation / emission Filter nomenclature • Excitation filters: x • Emission filters: m • Beamsplitter (dichroic mirror): bs, dc, FT • 480/30 = the center wavelength is at 480nm; full bandwidth is 30 [ = +/- 15] • BP = bandpass, light within the given range of wavelengths passes through (BP 450-490) • LP = indicates a longpass filter which transmits wavelengths longer than the shown number and blocks shorter wavelengths (LP 500) • SP = indicates a shortpass filter which transmits wavelengths shorter than the shown number, and blocks longer wavelengths Excitation / emission excitation and emission spectra of EGFP (green) and Cy5 (blue) excitation and emission spectra of EGFP (green) and Cy2 (blue) No filter can separate these wavelengths! Where to check spectra? You can plot and compare spectra and check spectra compatibility for many fluorophores using the following Spectra Viewers. Invitrogen Data Base BD Fluorescence Spectrum Viewer University of Arizona Data Base NCI ETI Branch flow Cytometry Standard techniques • wide-field • confocal • 2-photon Wide-field fluorescence • reflected light method • Multiple wavelength source (polychromatic, i.e. mercury lamp) • Illumination of whole sample upright Zeiss microscopes, fluorescence tissue culture microscopes, timelapse microscopes PFS timelapse • New long term timelapse (Nikon) • System adjusts the focus by using IR laser to measure the distance to the glass of your dish Wide-field vs confocal Wide-field image confocal image Molecular probes test slide Nr 4, mouse intestine Confocal • method to get rid of the out of focus light less blur • whole sample illuminated (by scanning single wavelength laser) • only light from the focal plane is passing through the pinhole to the detector Confocal Use: • to reduce blur in the picture high contrast fluorescence pictures (low background) • optical sectioning (without cutting); 3D reassembly possible Careful: increasing image size (more pixels) does not mean that the objective can resolve the same!!! (resolution determined by NA, a property of the objective) Timelapse with confocal You can do timelapse movies with the confocal. Mainly for fast processes Be aware that not all our confocals have incubation chamber and CO2! Two Leica confocals and one Olympus FV 1000 2-photon microscopy Excited state Excitation: long wavelength (low energy) Emission: shorter wavelength (higher energy) than excitation Each photon gives ½ the required energy Ground state 2-photon microscopy Use of lower energy light to excite the sample (higher wavelength) 1-photon: 488nm 2-photon: 843nm Advantages: IR light penetrates deeper into the tissue than shorter wavelength 2-photon excitation only occurs at the focal plane less bleaching above and below the section Use for deep tissue imaging new La Vision microscope (live mouse imaging, will be installed in the new building) Special applications: • FRET and FLIM • FRAP and photoactivation • TIRF FRET (Fluorescence Resonance Energy Transfer) • method to investigate molecular interactions • Principle: a close acceptor molecule can take the excitation energy from the donor (distance ca 1-10 nm) No FRET Exited state FRET situation: Excitation of the donor (GFP) but emission comes from the acceptor (RFP) Exited state Energy transfer, no emission! Exited state Donor (GFP) Acceptor (RFP) Ground state Ground state Ground state FRET ways to measure: • Acceptor emission Detect the emission of the acceptor after excitation of the donor, e.g. excite GFP with 488 but detect RFP at 610 (GFP emission at 520) • Donor emission after acceptor bleaching take image of donor, then bleach acceptor (with acceptor excitation wavelength - RFP:580nm), take another image of donor should be brighter! FRET You need: • a suitable FRET pair (with overlapping excitation/emission curves) Disadvantages: • Bleed through (because of overlapping spectra) Limitation of techniques (filters etc) • Photobleaching only with fixed samples • Intensity depends on concentrations etc FLIM (Fluorescence Lifetime Imaging Microscopy) • measures the lifetime of the excited state (delay between excitation and emission) • every fluorophore has a unique natural lifetime • lifetime can be changed by the environment, such as: Ion concentration Oxygen concentration ∆t=lifetime pH Protein-protein interactions FLIM Lifetime histogram Excitation of many electrons at the same time count the different times when they are falling back down (i.e. photons are emitted) decay curve lifetime = ½ of all electrons are fallen back Example of FLIM-FRET measurement GFP expressed in COS 1 cell: average lifetime of 2523 ps fused GFP-RFP expressed in COS 1 cell: average lifetime of 2108 ps Joan Grindlay, R7 FLIM You still need: a suitable FRET-pair with the right orientation of the π-orbitals Interaction of proteins is not enough, because fluorophores have to be close enough and in the right orientation! Use of FLIM: measurements of concentration changes (Ca2+), pH change etc, Protein interactions FRET: Leica confocal 2 or Olympus FV 1000 FLIM: Leica confocal 1 and soon LIFA system from Lambert Instruments Special applications: • FRET and FLIM • FRAP and photoactivation • TIRF FRAP (Fluorescence Recovery After Photobleaching) • Intense illumination with 405 laser bleaches the sample within the selected region observation of the recovery before 0.65 s 0.78 s Use: to measure the mobility/dynamics of proteins under different conditions Olympus FV 1000 photoactivation • Fluorophore only becomes active (= fluorescent) if excited (e.g. with 405 laser) due to structural change Pictures taken from a activation movie: activation of a line trough the lamellipodia of the cell, activated GFP_F diffuses quickly Olympus FV 1000 Special applications: • FRET and FLIM • FRAP and photoactivation • TIRF TIRF (Total Internal Reflection Fluorescence) You need: • TIRF objectives with high NA • TIRF condensor, where you are able to change the angle of illumination • Glass coverslips TIRF micro.magnet.fsu.edu Result: very thin section at the bottom of the sample 150-200nm Use: to study membrane dynamics (endocytosis, focal adhesions, receptor binding) Nikon TE 2000 TIRF vs epi FAK-lasp in epi mode (wide field) FAK-lasp in tirf mode (wide field) Heather Spence, R10 TIRF vs epi Lasp in confocal sectioning Lasp in TIRF mode Heather Spence, R10 Summary/comparison method excitation detection sectioning Wide field Whole sample Whole sample No sectioning Simple fluorescence samples confocal Whole sample One z-plane 350-500nm High contrast images, optical sectioning 2-Photon One z-plane One z-plane 500-700nm Deep tissue imaging, optical sectioning FLIM/FRET FRAP + photoactivation TIRF use Protein interactions dynamics/mobility 405 laser (UV) Only bottom plane Only bottom plane 150-200nm Membrane dynamics • Please book proper training with Tom or Margaret before using BAIR equipment! BAIR webpage demonstration: