Laboratory #2
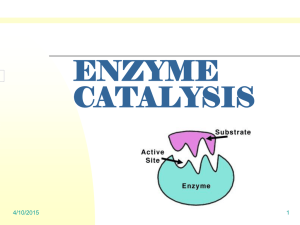
Enzyme Catalysis
[Name]
[Period]
[Date Performed]
Abstract
The Effect of Enzyme Catalysis Concentration on the Substrate Reaction Rates was studied by measuring the
effects of Catalase enzymes on hydrogen peroxide decomposition. The rate of the reaction was determined by
measuring the rate of the reaction when hydrogen peroxide and Catalase are mixed at the same ratio for
different time (10, 20 30 60 120 180 360 seconds).
Introduction:
Enzymes are catalytic proteins, meaning they speed up – but do not create – chemical reactions, without being
used up or altered permanently in the process. Although various enzymes use different methods, all accomplish
catalysis by lowering the free energy of activation – activation energy – for the reaction, thus allowing it to
occur more easily.
Enzymes employ a variety of methods for performing catalysis. Some provide a micro environment within the
active site where some of the side chains are H+ or OH- donors or receivers. Other enzymes work by bringing
together substrates that would not normally meet outside the enzyme, or orienting them in a manner in which
they would otherwise not occur. Still other enzymes stress the bonds of substrate molecules in order to make
them easier to break; some take this a step further by actually forming temporary covalent bonds with the
substrate molecules. Regardless of how it is done, all enzyme catalyzed reactions are reversible and will turn
around when necessary.
Each enzyme has a unique and highly complex structure, which is given rise to through the four levels of
protein organization. The specific arrangement of the amino acids via polypeptide bonds is the primary structure
of the protein. Formed by ribosomes and encoded in mRNA, the primary structure gives rise to all other levels
of organization. Secondary structure is formed of α-helices and β-pleated sheets which are formed when
twisting and folding polypeptides form hydrogen bonds along the nitrogenous backbone of the protein. The
third level of organization, tertiary structure, consists of irregular loops and folds which form as the protein
folds and are held in place be disulfide bridges and hydrophobic interactions. Disulfide bridges are strong
covalent bonds that occur between the sulfurs of various amino acids; hydrophobic interactions occur when
large numbers of hydrophobic side chains come together and ‘stick’ to one another. Quaternary structure, the
fourth and final level of protein organization, occurs only when multiple polypeptides come together in a single
protein. This structure is the final overall three-dimensional shape of the protein.
As a result of four levels of organization, an enzyme has a very specific shape, which is called its conformation.
Even more specific is the active site of the enzyme, where the actual catalysis occurs. The specific molecule or
closely related molecules on which an enzyme functions is known as its substrate. Shape plays such an
important role in enzymatic catalysis, that often even isomers of the substrate will be rejected. Once the
substrate enters the active site, it may begin a process known as induced fit in which the enzyme perfectly
conforms to the molecule to allow for more efficient catalysis.
Enzymes have specific environmental conditions at which they will function best. As a result, changes in
environment can severely impact enzyme catalysis in both negative and positive ways. Most enzymes found in
the human body, as is catalase, are designed to work best in the range of 35-40°C and a pH of 6-8; those in heat
loving thermopiles can operate in excess of 70°C. Each enzyme has specific ranges at which it optimally
functions; in general, increasing the temperature will help the reaction along, until the point at which the protein
degrades and denatures – or falls apart into its lower level structures. Denatured proteins will often return to
their original state, after the removal of the denaturing agent, except when they are degraded multiple levels
(such as Quaternary to Secondary). Rate of reaction through catalysis can also be increased by increasing the
concentration of either the enzyme or the reactants; enzyme if all the active sites are full or the substrate if the
active sites are not all full. In this exercise the reaction will begin rather quickly with the addition of the
enzyme, but will slow as the H2O2 is converted to H2O and O2. I also believe that, given the amount of time the
reactions are to be run for, not all of the H2O2 will react, thus leaving some to react with the KMnO4 even after
the full 360 seconds.
Methodology:
Materials
Exercise 2A
In Part 1, 10 mL of 1.5% of H2O2, a 50 mL beaker, and 1 mL of catalysis are needed. In Part 2, 5mL of
catalysis, a water bath, and 10 mL of 1.5% H2O2 is needed. In Part 3, a 1 cm³ of liver, 50 mL beaker, and 10 mL
of 1.5% H2O2. For all three parts, safety goggles, lab aprons, pencil, paper, erasers, and paper towels are needed.
Exercise 2B
To do this experiment, 10 mL of 1.5% H2O2, 1 mL of water, 10 mL of H2SO4, 50 mL beaker, 25 mL beaker, 5
mL syringe, and KmnO4 are needed. Safety goggles, lab aprons, pencil, paper, erasers, and paper towels are also
needed.
Exercise 2C
To do this exercise, safety goggles, lab aprons, pencil, paper, erasers, paper towels, about 20 mL of 1.5% H2O2,
1 mL of H2O, 10 mL of H2SO4, 50 mL beaker, 25 mL beaker, 5 mL syringe, and KmnO4 are needed.
Exercise 2D
To do this experiment, about 60 mL of 1.5% H2O2, 6 mL of catalysis, 60 mL of H2SO4, 12 cups labeled 10, 30,
60, 120, 180, and 360 seconds, six cups labeled acid, a black marker, and a timer are needed. Safety goggles,
lab aprons, pencil, paper, erasers, and paper towels will be needed, also.
Procedure
Exercise 2A
In Part 1, transfer 10 mL of 1.5% H2O2 a 50 mL glass beaker and add 1 mL of freshly made catalase to the
solution. Remember to keep the catalase solution on ice at all times. Record the observations made. In Part 2,
transfer 5 mL of the purified catalase extract to a test tube and place it in a boiling water bath for five minutes.
Next, transfer 10 mL of 1.5% H2O2 into a 50 mL beaker and add 1 mL of the cooled, boiled catalase solution.
Again record the results. In Part 3, cut 1 cm of liver, transfer it into a 50 mL glass beaker containing 10 mL of
1.5% H2O2, and mix it. Record the results.
Exercise 2B
To form a baseline for this experiment, put 10 ml of 1.5% H2O2 into a clean glass beaker. Add 1 mL of H2O and
then add 10 mL of H2SO4 (1.0 M). Be careful when using acid. Mix this solution well. Remove a 5 mL sample
and place it into another beaker. Assay for the amount of H2O2 as follows. Place the beaker containing the
sample over white paper and use a 5 mL syringe to add one drop of KMnO4 at a time to the solution until it
becomes a persistent pink or brown color. Gently swirl the solution after adding each drop. Record all results.
Exercise 2C
To determine the rate of spontaneous conversion of H2O2 to H2O and O2 in an uncatalyzed reaction, put about
20 mL of 1.5% H2O2 in a beaker. Store it uncovered at room temperature for approximately 24 hours. Put 10 ml
of 1.5% H2O2 into a clean glass beaker (using the uncatalyzed H2O2 that set out). Add 1 mL of H2O and then
add 10 mL of H2SO4 (1.0 M). Be careful when using acid. Mix this solution well. Remove a 5 mL sample and
place it into another beaker. Assay for the amount of H2O2 as follows. Place the beaker containing the sample
over white paper and use a 5 mL syringe to add one drop of KMnO4 at a time to the solution until it becomes a
persistent pink or brown color. Gently swirl the solution after adding each drop. Record all results.
Exercise 2D
If a day or more has passed since Exercise B was performed, it is necessary to reestablish the baseline. Repeat
the assay from Exercise B and record the results. Compare with other groups to check that results are similar.
To determine the course of an enzymatic reaction, how much substrate is disappearing over time must be
measured. The first thing to be done is to set up the cups labeled with times and acid. Add 10 mL of H2SO4 to
each of the cups marked acid. Then put 10 mL of 1.5% H2O2 into the cup marked 10 sec. Add 1 mL of catalase
extract to this cup. Swirl gently for 10 seconds (use the timer for accuracy). At 10 seconds, add the contents of
one of the acid filled cups. Remove 5 mL and place in the second cup marked 10 sec. Assay the 5 mL sample
by adding one drop of KMnO4 at a time until the solution turns a pink or brown. Repeat the above steps except
allow the reactions to proceed for 30, 60, 120, 180, and 360 seconds, respectively. Use the times corresponding
with the marked cups
Results:
Table 1
Table 2
Catalysis Activity
Experiment
Observations
H2O2 and Fresh Catalase
Small amount of bubbles
H2O2 and Boiled Catalase
Little or no bubbling
Catalase with Liver
Very bubbly or reactive
Baseline Assay
Baseline Calculations
Table 3
Final Reading of Burette
1.5 mL
Initial Reading of Burette
5.0 mL
Baseline (Final-Initial)
3.4 mL of KmnO4
The Uncatalyzed Rate of H2O2 Decomposition
Final Reading of Burette
2.2 mL
Initial Reading of Burette
7.0 mL
Amount of KMnO4 Titrant
4.8 mL
H2O2 Spontaneously Decomposed
1.3 mL
Percentage Spontaneously Decomposed in 24 hours
62.9%
Table 4
New Baseline
Baseline Calculations
Table 5
Final Reading of Burette
1.4 mL
Initial Reading of Burette
5.0 mL
Baseline (Final-Initial)
3.6 mL of KmnO4
Catalyzed Rate of H2O2 Decomposition
Time (seconds)
Graph 1
10
30
60
120
180
360
A. Baseline
3.6 mL
3.6 mL
3.6 mL
3.6
mL
3.6 mL
3.6
mL
B. Final
Reading
1.2 mL
1.4 mL
1.8 mL
1.9
mL
2.4 mL
2.8
mL
C. Initial
Reading
5 mL
5 mL
5 mL
5
mL
5 mL
5
mL
D. Amount of
KmnO4
Consumed
(B-C)
3.8 mL
3.6 mL
3.2 mL
3.1
mL
2.6 mL
2.2
mL
E. Amount of
H2O2 Used
(A-D)
.2 mL
0 mL
.4 mL
.5
mL
1.0 mL
1.4
mL
Affect of Time on Enzyme-Catalyzed H2O2 (Remaining amount)
Error Analysis:
Any errors occurring in this experiment could have been caused many factors. Most common are
Miscalculations involving experimenter error when observing, measuring and gathering data; that is, errors by
misreading of a syringe, miscalculating the data on the tables, or with the inadvertent mixing of reagents. There
is also the probability or errors as a result of the materials; for example, when calculating the percent of
hydrogen peroxide spontaneously decomposed after 24 hours, a relatively new hydrogen peroxide reagent
would yielded a much different percentage of oxygen molecules than an aged hydrogen peroxide. These error
could have been minimized with more trials and an average. In Exercise D especially, the factor of planning
became increasingly essential. The first attempt at 2D was unsuccessful due to several reasons. First of all, the
measurements, which were taken, could have possibly been inaccurate and the 60-mL syringe containing H2SO4
also dripped into one of the cups early which did not allow the reaction to fully take place. There was also some
confusion on the operation of the timer and precise planning in its use. The second attempt at 2D contained
errors as well. The measurements were still not as accurate as they should have been, and the solution did not
appear entirely uniform. In one cup, for example, the first drop of KMnO4 left a persistent pink color, and then
after over a minute, it returned back to being clear. It then took several milliliters more to get it back to a pink
color
Conclusion:
The results were a bit of a surprise to me. First of all, I would not have expected the reaction to occur quite so
quickly. Secondly, I did not expect the amount of H2O2 to get to the point where 1 drop (~0.05mL) of KMnO4 is
enough to stain 5mL of reacted solution. In that regard, I am drawn to believe that we would require more
accurate instruments to acquire anything like the ‘perfect’ data; otherwise it may have been better for each
group to perform the full range of the experiment.
As for my group’s results, I believe we went wrong in establishing our baseline – it did seem as though 1.8 was
a little low and this was confirmed by the lab director. In my opinion I feel one of two things may have
happened, either we had some impurity in our beaker or we misread the markings on the burette; sadly, the later
seems more probable. It is, however, possible that something was contaminated in our burette as well, as
something did not look quite right when we were setting everything up.
The rates of reaction beyond the sheer speed were not much of a surprise; but some stand out in my mind;
namely the difference in rate during the first two measurements. From 0 seconds to 10 seconds, the rate of
reaction averaged .13mL H2O2 decomposed per second; for 10 seconds to 30 seconds, the rate fell to a mere
.0075mL H2O2 decomposed per second – approximately a 95 percent decrease in only 20 seconds!
Accuracy was obviously a problem with the class results, given the fact that three of eight groups had data
blatantly inconsistent with the general trend and the perfect data. I, however, must partake in some of the blame
as my group was one of the three – although I contend we preformed the experiment correctly and only messed
up on the baseline – which had inconsistent data. It should be noted, however, that this was our first lab of the
year and no one can blame us for such a mistake.
My hypothesis was partially correct as the reaction most certainly did begin quickly and slow as the time moved
on, but I was essentially (for our intents and purposes) incorrect in the idea that enough H2O2 would remain so
that the KMnO4 would react with something. While I am sure some remained, though the amount may have
been very minute, it was simply not enough to notice at the scale we were using. It is my firm belief that, given
micro liter quantities of KMnO4, I would have been able to assay the presence of some H2O2 remaining in the
solution. Overall, however, my hypothesis was only partially correct.
References:
AP® Biology Lab Manual for Students (2001 Revised). Laboratory #2 Enzyme Catalysis. (New Jersey, USA)
Carolina Scientific® Advanced Placement Biology Laboratory Manual for Students (2005). Laboratory #2
Enzyme Catalysis. (North Carolina, USA)
Mader, S. Biology. 9th ed. New York: The McGraw-Hill Companies, 2007.
Questions:
REWRITE AND ANSWERS QUESTIONS HERE
 0
0