Supplementary Information (doc 52K)
advertisement
")
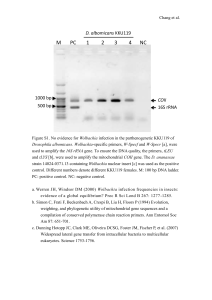
Legends Supplementary Material Table S1 This table contains a simulated community containing 3 taxa (taxon 1-3) in two different treatment (treatment x,y) in order to demonstrates how PP, TP, community functional change and theoretical community functional change were calculated. We constructed communities for 2 different scenarios: 1) no replacement of functions among individual taxa, which results in identical values for the community functional change and theoretical community functional change, and 2) replacement of functions among individual taxa, which results in a lower value for the community functional change compared to the theoretical community functional change and therefore indicates functional stability. The degree of functional stability in the second scenario is determined by the difference between the theoretical community functional change and the community functional change. Table S2 List of taxa used for calculations in the manuscript. For each taxon, information on the phylogeny and abundance within treatments, average abundance of both treatments within an experiment, fold-difference, PP (Bray-Curtis dissimilarities of gene composition of size normalized taxa), BCfd (Bray-Curtis dissimilarities of gene composition of non size-normalized taxa) and TP (BCfd corrected for the change of sample size in non-size-normalized samples) is given. We further provide results from a Blastn search of 16S rRNA gene sequences of the representative genomes against 16S rRNA gene OTUs sequenced in previous studies from the same samples (184 and 284 OTUs available for Exp_SwDi and Exp_CyDi; Landa et al. 2013a; Landa et al. 2013b). The 16S rRNA OTUs (410 bp) contain the variable regions V2/V3 and reported similarity values to 16S rRNA gene sequences of reference genomes underestimate similarities for the full length 16S rRNA gene. (Query ID: Genbank ID for DNA sequence containing the 16S rRNA gene of the reference genomes; Subject ID: ID for 16S rRNA OTUs from our samples). We also show the bin-specific fractions of transcripts coding for ribosomal proteins (KEGG pathway ko03010), proteins involved in oxidative phosphorylation (KEGG pathway ko00190), and RNA polymerase rpoB (KEGG number K03043). These housekeeping genes should be present and transcribed in all members of a community actively growing in continuous culture via aerobic metabolism. A single taxon bin that did not express transcripts coding for oxidative phosphorylation may be the exception, potentially suggesting an alternate niche for anaerobic growth (e.g., embedded in particulate material). Missing transcripts in some taxon bins for the rpoB gene may be caused by low abundance of the taxon or binning errors. Figure S1 To check the consistency with which metatranscriptomic data was assigned to genome bins, we compared the transcriptome composition of 5 highly abundant taxa (minimum 5% of all transcripts in at least one biological replicate) after normalizing to the same number of reads by subsampling. Transcripts were subsequently clustered using Bray-Curtis dissimilarity and Ward’s linkage. (Alc: Alcanivorax sp. DG881, Col: Colwellia psychrerythraea, Gla: Glaciecola sp. HTCC2999, Nep: Neptuniibacter caesariensis, IMC: gammaproteobacterium IMCC1989; for abbreviation for cultures, see Table 1). Transcriptome bins from Exp_SwDi are labeled with filled circles whereas those from Exp_CyDi are labeled with open circles. The dendrogram demonstrates that each reference genome bin was more similar in transcript composition across different replicates and treatments than when compared to the other genome bins. The results provide evidence that at least among treatments within one experiment, transcripts binning to a taxon were derived from genetically related organisms. Figure S2 Percent protein identity of transcripts to the reference genome for universal singlecopy genes rpoB (green) and recA (blue) for the three most abundant taxa in communities of the control/cyano treatment (Exp_SwDi/Exp_CyDi, dashed line) and diatom treatment (Exp_SwDi and Exp_CyDi, solid line). A single peak indicates that a single organism was sampled for the gene and overlapping peaks for the same gene across treatments indicates that sequences originating from highly related populations were being compared. Figure S3 Plot of PP versus the average abundance of taxa within each experiment. Regression analysis suggests that the magnitude of PP is independent of taxon abundance (r2: 0.007, p:0.319). Introducing a weighting factor for taxon abundance therefore did not introduce a bias when calculating the average weighted PP/TP. Figure S4 Distribution of taxon bin-specific fractions of transcripts coding for ribosomal proteins (KEGG pathway ko03010), oxidative phosphorylation (KEGG pathway ko00190), and RNA polymerase rpoB (KEGG number K03043). Individual taxa with high fractions of the targeted genes may reflect unique transcription patterns in this taxon, but could also indicate binning errors.