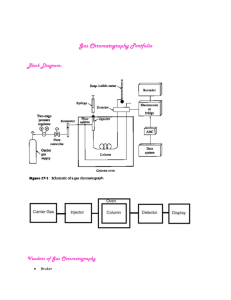
solid chromatography
advertisement

CHROMATOGRAPHIC METHODS Chromatography is a method of separating a mixture into its parts and identifying those parts. Analysis of complex mixtures requires separation and isolation of components. Chromatography can separate very complex mixtures composed of many very similar components. IUPAC defines chromatography as a “physical method of separation in which the components to be separated are distributed between two phases, one of which is stationary and the other moves in a definite direction”. Chromatographic methods make use of a mobile phase and a stationary phase. Common characteristics (or requirements) of chromatographic methods are given below. 1) The stationary and mobile phases should be immiscible. 2) An arrangement is required to deposit the mixture at one end of the stationary phase. 3) The mobile phase is allowed to flow to the other end of the stationary phase. This flow of mobile phase is called elution. The mobile phase is also known as eluent. 4) When the mobile phase passes through the stationary phase the components of the mixture partition between the mobile and stationary phases. Rate of partitioning will be different for different components of the mixture. The partitioning process is repeated to get a near complete separation. The component having the lowest partition coefficient gets separated at first. 5) The separated components are visualized as spots or bands or are detected by using special instruments. The mobile phase is usually a liquid or a gas and the stationary phase is usually a solid or a liquid film coated on a solid surface. Chromatographic methods can be classified by the physical state of the mobile and stationary phases. Chromatographic methods are named by listing the type of mobile phase followed by the type of stationary phase. For example in gas-liquid chromatography, the mobile phase is a gas and the stationary phase is a liquid. If only one phase is given (for example gas chromatography) it will be the mobile phase. This classification involves three major classes, gas chromatography, liquid chromatography and supercritical fluid chromatography. Each of these can be subdivided based on the types of stationary phases involved. For example, in gas-liquid chromatography the stationary phase is a liquid whereas in gas-solid chromatography the stationary phase is a solid. Chromatographic methods can also be classified by the method of contact between the mobile phase and stationary phase. The stationary phase and the mobile phase can be made in to contact by two methods, column chromatography and planar chromatography. In column chromatography the stationary phase is placed in a narrow column through which the mobile phase moves under the influence of gravity or pressure. In planar chromatography the stationary phase is coated on a flat glass, metal or paper and it is placed in a developing chamber containing the mobile phase. The mobile phase moves by capillary action. Third classification of chromatographic methods is by the mechanism by which the components of the sample get separated. In adsorption chromatography, separation is based on the ability of the components to get adsorbed on a solid stationary phase. In partition chromatography separation is based on the difference in partition coefficients of the components. In ion-exchange chromatography, the stationary phase contains anionic (e.g. -SO3-) or cationic (e.g. –N(CH3)3+) groups. Ionic components are attracted towards the stationery phase by electrostatic force of attraction. Adsorption Chromatography In adsorption chromatography the separating mechanism is adsorption. Generally in liquid-solid chromatography (LSC) a polar stationary phase and a non polar mobile phase is used. This is called normal phase LSC (NPLSC). In some cases a non polar stationary phase and polar mobile phase is used and this method is called reverse phase LSC (RPLSC). Most commonly used stationary phases are silica and alumina and the stationary phase should be completely dry. For RPLSC charcoal based adsorbents are used as stationary phase. Commonly used mobile phases include hexane, isooctane, dichloromethane, acetone, ethyl acetate, chloroform, ethanol etc. In column chromatographic method, the stationary phase is packed in a long column and the mixture to be separated is deposited on top of the stationary phase. Components of the sample get adsorbed on the stationary phase and the extent of adsorption will be different for different components depending on the polarity of components. Mobile phase is continuously passed through the stationary phase and this process is called elution. When the mobile phase flows through the stationary phase, there is a competition between adsorption versus dissolution. Weakly adsorbed components easily get dissolved in the mobile phase. In NPLSC, since a polar stationary phase is used, polar components get strongly adsorbed on the stationary phase. Hence the first compound to reach the detector will be the least polar compound. Finally a detector placed at the end of the column responds to the concentration of analytes. In column adsorption chromatography, the efficiency and speed of the method depends on a number of factors such as surface area of the stationary phase, column height, diameter of the column etc. The efficiency will be increased if surface area of the stationary phase is more, i.e., when smaller particles are used. But when smaller particles are used time taken for the analysis will be more. Also efficiency of the process increases with increase in column height and decrease in diameter of the column. But both these factors increase the time required for the process. For many samples liquid-liquid chromatography (LLC) is superior to LSC. The sensitivity of LLC is more compared to LSC. Therefore for the separation of a sample containing closely related compounds LLC is more suitable. Partition Chromatography In partition chromatography the mobile phase can be either a liquid or a gas and the stationary phase is a liquid. In liquid-liquid partition chromatography, the sample solvent is the stationary phase and the extracting solvent is the mobile phase. When the mobile phase passes through the stationary phase; the components of the sample partition or distribute themselves between the two phases. The separation is based on the solubilities of the components in the two phases or the polarity of the components. If the stationary phase is polar, it will retain polar components more than it will non polar components. Thus nonpolar components will move quickly through the stationary phase than polar components. The condition for partition chromatography is that the stationary phase and the mobile phase should be immiscible. In normal phase partition chromatography, the stationary phase is a polar solvent like water and the mobile phase will be a non polar solvent. In reverse phase partition chromatography the stationary phase is non polar and mobile phase is polar. But the stationary phase will always be slightly soluble in the mobile phase. So after some time the stationary phase will be carried away by the mobile phase. In practice, in order to avoid this problem, a liquid chemically bonded to the surface of a solid support is used as the stationary phase. In partition chromatography, the mobile phase can also be a gas. In this case the relative solubilities of the components in the mobile phase are not important; instead the relative vapour pressures of the components in the mobile phase are important. High-Performance Liquid Chromatography (HPLC) Although gas chromatography is widely used, it is limited to samples that are thermally stable and easily volatilized. Nonvolatile samples, such as peptides and carbohydrates, can be analyzed by GC, but only after they have been made more volatile by a suitable chemical derivatization. For this reason, the various techniques included within the general scope of liquid chromatography are among the most commonly used separation techniques. In HPLC, a liquid sample, or a solid sample dissolved in a suitable solvent, is carried through a chromatographic column by a liquid mobile phase. Separation is determined by solute/stationary-phase interactions, including liquid–solid adsorption, liquid–liquid partitioning, ion exchange and size exclusion, and by solute/mobile-phase interactions. In each case, however, the basic instrumentation is essentially the same. A schematic diagram of a typical HPLC instrument is shown below. HPLC Columns An HPLC typically includes two columns: an analytical column responsible for the separation and a guard column. The guard column is placed before the analytical column, protecting it from contamination. The most commonly used analytical columns for HPLC are constructed from stainless steel with internal diameters between 2.1 mm and 4.6 mm, and lengths ranging from approximately 30 mm to 300 mm. These columns are packed with 3–10 μm porous silica particles that may have an irregular or spherical shape. Microcolumns use less solvent and, because the sample is diluted to a lesser extent, produce larger signals at the detector. These columns are made from fused silica capillaries with internal diameters of 44–200 mm and lengths of up to several meters. Open tubular micro columns also have been developed, with internal diameters of 1–50 μm and lengths of approximately 1 m. These columns contain no packing material. Two problems tend to shorten the lifetime of an analytical column. First, solutes binding irreversibly to the stationary phase degrade the column’s performance by decreasing the available stationary phase. Second, particulate material injected with the sample may clog the analytical column. To minimize these problems, a guard column is placed before the analytical column. Guard columns usually contain the same particulate packing material and stationary phase as the analytical column, but are significantly shorter and less expensive; a length of 7.5 mm and a cost one-tenth of that for the corresponding analytical column is typical. Because they are intended to be sacrificial, guard columns are replaced regularly. Stationary Phases In liquid–liquid chromatography the stationary phase is a liquid film coated on a packing material consisting of 3–10 μm porous silica particles. The stationary phase may be partially soluble in the mobile phase, causing it to “bleed” from the column over time. To prevent this loss of stationary phase, it is covalently bound to the silica particles. Bonded stationary phases are attached by reacting the silica particles with an organochlorosilane of the general form Si(CH3)2RCl, where R is an alkyl or substituted alkyl group. To prevent unwanted interactions between the solutes and any unreacted –SiOH groups, the silica frequently is “capped” by reacting it with Si(CH3)3Cl; such columns are designated as end-capped. The properties of a stationary phase are determined by the nature of the organosilane’s alkyl group. If R is a polar functional group, then the stationary phase will be polar. Examples of polar stationary phases include those for which R contains a cyano (–C2H4CN), diol (–C3H6OCH2CHOHCH2OH), or amino (– C3H6NH2) functional group. Since the stationary phase is polar, the mobile phase is a nonpolar or moderately polar solvent. The combination of a polar stationary phase and a nonpolar mobile phase is called normal-phase chromatography. In reverse-phase chromatography, which is the more commonly encountered form of HPLC, the stationary phase is nonpolar and the mobile phase is polar. The most common nonpolar stationary phases use an organochlorosilane for which the R group is an n-octyl (C8) or n-octyldecyl (C18) hydrocarbon chain. Most reverse-phase separations are carried out using a buffered aqueous solution as a polar mobile phase. Because the silica substrate is subject to hydrolysis in basic solutions, the pH of the mobile phase must be less than 7.5. Mobile Phases The elution order of solutes in HPLC is governed by polarity. In a normal-phase separation the least polar solute spends proportionally less time in the polar stationary phase and is the first solute to elute from the column. Retention times are controlled by selecting the mobile phase, with a less polar mobile phase leading to longer retention times. In a reverse-phase separation the order of elution is reversed, with the most polar solute being the first to elute. Increasing the polarity of the mobile phase leads to longer retention times, whereas shorter retention times require a mobile phase of lower polarity. Isocratic Versus Gradient Elution:- When a separation uses a single mobile phase of fixed composition it is called an isocratic elution. It is often difficult, however, to find a single mobile-phase composition that is suitable for all solutes. A mobile phase that is suitable for early eluting solutes may lead to unacceptably long retention times for later eluting solutes. Optimizing conditions for late eluting solutes, on the other hand, may provide an inadequate separation of early eluting solutes. Changing the composition of the mobile phase with time provides a solution to this problem. For a reverse-phase separation the initial mobile-phase composition is relatively polar. As the separation progresses, the mobile phase’s composition is made less polar. Such separations are called gradient elutions. HPLC Plumbing An important feature of HPLC instrumentation is the presence of several solvent reservoirs. The availability of several solvent reservoirs allows the mobile phase’s composition to be quickly and easily varied. This is essential when using a gradient elution. Before they are used, mobile-phase solvents must be treated to remove dissolved gases, such as N2 and O2, and small particulate matter, such as dust. Dissolved gases often lead to the formation of gas bubbles when the mobile phase enters the detector, resulting in a distortion of the detector’s signal. The mobile-phase solvents are pulled from their reservoirs by the action of a pump. A solvent proportioning valve controls the mobile phase’s composition. The back and forth movement of a reciprocating pump results in a pulsed flow that contributes noise to the chromatogram. To eliminate this problem a pulse damper is placed at the outlet of the pump. Sample Introduction In HPLC, the sample is introduced using a loop injector. In the load position the sampling loop is isolated from the mobile phase and is open to the atmosphere. A syringe with a capacity several times that of the sampling loop is used to place the sample in the loop. Any extra sample beyond that needed to fill the sample loop exits through the waste line. After loading the sample, the injector is turned to the inject position. In this position the mobile phase is directed through the sampling loop, and the sample is swept onto the column. Detectors for HPLC As with gas chromatography, numerous detectors have been developed for use in monitoring HPLC separations. UV/Vis absorbance detectors These detectors monitor the absorption of UV or visible light by analytes in the HPLC eluent. A typical UV/Vis detector consists of a deuterium source and a monochromator (a movable grating controlled by stepper motor to select wavelength through an exit slit) to focus the light through a small flow cell. A dual-beam optical bench is typical for reducing drift. Two photodiodes are used to measure light intensities of sample and reference beams. Advantages:- compound-specific; nondestructive; concentration detector; compound sensitivities differ over a wide range; useable with isocratic or gradient mobile phases including buffer salts; pre-, or post-column derivatization can be used to increase number of measurable compounds. Fluorescence (Fl) detector A fluorescence detector monitors the emitted fluorescent light of the HPLC eluent. It is selective and extremely sensitive (pg to fg) to highly fluorescent compounds. A detector consists of a xenon source, an excitation monochromator, an emission monochromator, a square flow cell, and a photomultiplier for amplifying the emitted photons. A pulsed xenon source is becoming popular because it requires less power, has more energy in the far UV region, and allows detection modes such as phosphorescence, chemiluminescence, and bioluminescence. Refractive index (RI) detector Measuring a change in the mobile phase’s refractive index is analogous to monitoring the mobile phase’s thermal conductivity in gas chromatography. A refractive index detector is nearly universal, responding to almost all compounds, but has a poorer detection limit of 100 ng–1 mg of injected analyte. Furthermore, a refractive index detector is not useful for a gradient elution unless the mobile-phase components have identical refractive indexes. Electrochemical Detectors Another common group of HPLC detectors are those based on electrochemical measurements such as amperometry, voltammetry, coulometry, and conductivity. In amperometric detector effluent from the column passes over the working electrode, which is held at a potential favorable for oxidizing or reducing the analytes. The potential is held constant relative to a downstream reference electrode, and the current flowing between the working and auxiliary electrodes is measured. Detection limits for amperometric electrochemical detection are 10 pg–1 ng of injected analyte. Mass spectrometer Detection limits are quite good, typically 100 pg–1 ng of injected analyte, with values as low as 1–10 pg in some situations. In addition, a mass spectrometer provides qualitative, structural information that can help identify the analytes. The interface between the HPLC and mass spectrometer is technically more difficult than that in a GC–MS because of the incompatibility of a liquid mobile phase with the mass spectrometer’s high vacuum requirement. Evaporative light scattering detector (ELSD) An ELSD converts the HPLC eluent into a particle stream and measures the scattered radiation. It offers universal detection for nonvolatile or semivolatile compounds and has higher sensitivity than the RI detector (in the low ng range) in addition to being compatible with gradient analysis. ELSD consists of a nebulizer equipped with a constant temperature drift tube where a counter-current of heated air or nitrogen reduces the HPLC eluent into a fine stream of analyte particles. A laser or a polychromatic beam intersects the particle stream, and the scattered radiation is amplified by a photomultiplier. MODES OF SEPARATION IN HPLC Different modes of separation involved in HPLC are mentioned below. (i) Adsorption:- Adsorption is involved in liquid –solid chromatography. (ii) Partition:- Partition is involved in liquid –liquid chromatography. (iii) Ion-exchange:- Ion-exchange chromatography is a form of liquid chromatography in which the stationary phase is an ion exchange resin. In ion-exchange chromatography (IEC) the stationary phase is a cross-linked polymer resin, usually divinylbenzene cross-linked polystyrene, with covalently attached ionic functional groups. The counter ions to these fixed charges are mobile and can be displaced by ions that compete more favorably for the exchange sites. (iv) Size-Exclusion (v) Affinity (vi) Chiral Chromatography Applications of HPLC HPLC is routinely used for both qualitative and quantitative analyses of environmental, pharmaceutical, industrial, forensic, clinical, and consumer product samples. In quantitative analysis samples in liquid form can be analyzed directly, after a suitable clean-up to remove any particulate materials or after a suitable extraction to remove matrix interferents. Solid samples must first be dissolved in a suitable solvent, or the analytes of interest must be brought into solution by extraction. Gases are collected by bubbling through a trap containing a suitable solvent. Quantitative analyses are often easier to conduct with HPLC than GC because injections are made with a fixed-volume injection loop instead of a syringe. As a result, variations in the amount of injected sample are minimized, and quantitative measurements can be made using external standards and a normal calibration curve. SIZE-EXCLUSION CHROMATOGRAPHY (SEC) Size-exclusion chromatography is a form of liquid chromatography in which the stationary phase is a porous material and in which separations are based on the size of the solutes. Size-exclusion or gel chromatography is paI1icularly applicable to high-molecular weight species. Packings for sizeexclusion chromatography consist of small silica or polymer particles containing a network of uniform pores into which solute and solvent molecules can diffuse. While in the pores, molecules are effectively trapped and removed from the flow of the mobile phase. The average residence time of analyte molecules depends on their effective size. Molecules that are significantly larger than the average pore size of the packing are excluded and thus suffer no retention; that is, they travel through the column at the rate of the mobile phase. Molecules that are appreciably smaller than the pores can penetrate throughout the pore network and are thus entrapped for the greatest time; they are last to elute. Between these two extremes are intermediate-size molecules whose average penetration into the pores of the packing depends on their diameters. The fractionation that occurs within this group is directly related to molecular size and to some extent molecular shape. Note that size-exclusion separations differ from the other chromatographic procedures in the respect that no chemical or physical interactions between analyte and the stationary phase are involved. Some size exclusion packings are hydrophilic for use with aqueous mobile phases; others are hydrophobic and are used with nonpolar organic solvents. Chromatography based on the hydrophilic packings is sometimes called gel filtration chromatography (GFC), while techniques based on hydrophobic packings are termed gel permeation chromatography (GPC). With both types of packings, many pore diameters are available. The average molecular weight suitable for a given packing may be as small as a few hundred or as large as several million. Molecules above a given molecular weight are too large to enter the pores, and therefore elute in the void volume. Any molecule above this exclusion limit will elute in the minimum possible time, and molecules with molecular weights above this limit cannot be separated. Molecules small enough to freely diffuse into all portions of the pores will be the most strongly retained, spending the same portion of time trapped in the pores, so they will elute at the same maximum time, called the permeation limit, and will likewise fail to be separated. Thus in contrast to partition chromatography, SEC or GPC have not only a lower limit for peak retention times, but also an upper limit. This is actually an advantage, since chromatographic runs will not drag on too long with late eluters. In the molecular weight range between these limits (on the order of one to two orders of magnitude) the retention time of a molecule is inversely proportional to the logarithm of the molecular weight. A packing with the appropriate pore size to cover the anticipated range of weights must be selected. Particles with different pore sizes can be mixed to accomplish separation of mixtures of a wider range of molecular weights. Size-exclusion chromatography provides a rapid means for separating larger molecules, including polymers and biomolecules. Another important application is for the determination of formula weights. Since the retention volume is, to some degree, a function of a solute’s size and shape, reasonably accurate determinations of formula weight are possible only if the standards are carefully chosen to minimize the effect of shape. Size-exclusion chromatography can be carried out using conventional HPLC instrumentation, replacing the HPLC column with an appropriate size-exclusion column. A UV/Vis detector is the most common means for obtaining the chromatogram. CHIRAL CHROMATOGRAPHY Chiral chromatography is used for the separation of enantiomers. Either chiral mobile-phase additives or chiral stationary phases are required for these separations. Preferential complexation between the chiral resolving agent (additive or stationary phase) and one of the isomers results in a separation of the enantiomers. The chiral resolving agent must have chiral character itself to recognize the chiral nature of the solute. Generally chiral stationary phases are used. Here, a chiral agent is immobilized on the surface of a solid support. Several different modes of interaction can occur between the chiral resolving agent and the solute. In one type, the interactions are due to attractive forces such as those between πbonds, hydrogen bonds, or dipoles. In another type the solute can fit into chiral cavities in the stationary phase to form inclusion complexes. Affinity Chromatography Affinity chromatography involves covalently bonding a reagent called an affinity ligand, to a solid support. Typical affinity ligands are antibodies, enzyme inhibitors, or other molecules that reversibly and selectively bind to analyte molecules in the sample. When the sample passes through the column, only the molecules that selectively bind to the affinity ligand are retained. Molecules that do not bind pass through the column with the mobile phase. After the undesired molecules are removed. The retained analytes can be eluted by changing the mobile-phase conditions. The stationary phase for affinity chromatography is a solid such as agarose or a porous glass bead to which the affinity ligand is immobilized. The mobile phase in affinity chromatography has two distinct roles. First, it must support the strong binding of the analyte molecules to the ligand. Second, once the undesired species are removed. the mobile phase must weaken or eliminate the analyte-ligand interaction so that the analyte can be eluted. Often changes in pH or ionic strength are used to change the elution conditions during the two stages of the process. Affinity chromatography has the major advantage of extraordinary specificity. The primary use is the rapid isolation of biomolecules during preparative work. Paper Chromatography Paper chromatography makes use of a sheet of cellulose filter paper as the stationary phase. Since such paper is hydrophilic, the stationary phase is actually a thin film of water adsorbed on the surface of the paper. Thus, paper chromatography represents a form of partition chromatography. The mobile phase is always a liquid. The mixture to be separated is applied as a small spot within one inch of one end of a 10 inch filter paper. This method can also be used for the simultaneous separation of several mixtures. So on one paper nearly eight spots can be applied. The size of the spots should be small. After spotting, the paper is placed in a developing chamber containing a liquid mobile phase with the spotted end down. The mobile phase should lie below the level of the spots, i.e. they should not be in contact. The mobile phase moves upwards by capillary action and sweeps the spots along with it. While moving the components get separated from one another due to the difference in partition coefficient of different components. If the components are coloured then the spots can be visualized directly. If the components are not coloured, certain visualizing agents are used. Iodine is absorbed by most spots and makes them visible. Amino acids or proteins can be made visible by spraying ninhydrin. Alternatively spots can be made visible by placing the paper under UV light. Rf Value Paper chromatography can be used for qualitative analysis by calculating retardation factors (R f Values). These factors are based on the distance traveled by the mobile phase relative to the distance traveled by the components, each measured from the centre of the original spot. Rf value of a component can be given as, These factors which are less than or equal to one, can be compared with standard values to find out the identity of a compound. Rf values depend on a number of factors such as size and thickness of the paper, temperature, nature and quantity of mobile phase used, size of the developing chamber etc. If these factors are fixed, Rf values are reproducible and can be used for qualitative analysis. Rf value of a particular component can be compared with standard values to identify the component. Paper chromatography can be used for quantitative analysis also. The spot representing a particular component can be cut or scraped from the surface and dissolved in a suitable solvent. Quantitative analysis is done by using a suitable method like spectrophotometry, scanning densitometry etc. Paper chromatography is used for the separation and identification of a large number of compounds, especially biologically active compounds like amino acids, proteins, nucleic acids, drugs etc. In paper chromatography when the mobile phase is allowed to move upward, the technique is called ascending paper chromatography. In some cases descending paper chromatography is used in which the mobile phase is allowed to move downward. Radial Chromatography Radial chromatography is a special type of paper chromatography in which, a circular strip of paper is used instead of a rectangular paper. The circular paper contains a tongue or wick (a filter paper strip attached to the centre of the circular filter paper) at its centre. The mixture to be separated is spotted in the centre of the filter paper. The filter paper is placed on top of a petri dish which contains the mobile phase. The paper is placed such a way that the wick of the paper touches the mobile phase in the petri dish. The paper is covered with a glass plate. The mobile phase moves up through the wick, reaches the centre of the paper and spread in all directions. The components of the sample also move along with the mobile phase. Different components will appear not as spots but as concentric circles. Thin Layer Chromatography (TLC) In thin layer chromatography the stationary phase is coated uniformly as a thin layer on the surface of a rectangular glass, metal or plastic plate. For normal phase TLC either silica or alumina is used as the stationary phase. When silica and alumina are used the mechanism of separation is adsorption. Cellulose can also be coated on the surface of the plate. In this case water adsorbed on cellulose acts as stationary phase and the mechanism of separation is partition. The mobile phase in TLC is always a liquid. The mixture to be separated is spotted near one end of the plate and dried. The plate is kept in a developing chamber which contains the mobile phase. The level of the mobile phase should be below the spot. Mobile phase moves up by capillary action and takes the components along with it. Components of the sample get separated from one another by adsorption or partition. The separated components can be visualized as spots if the compounds are coloured. If the compounds are not coloured spots are made visible by using visualizing agents or UV light. Qualitative analysis can be done from the position of the spots. Rf values can also be used for qualitative analysis. But Rf values are less reproducible with TLC than with paper chromatography. For quantitative analysis the spot is carefully removed from the plate and dissolved in a suitable solvent and then analysis is done by a suitable method. TLC has a number of applications in laboratory and industry. It is used for monitoring the progress of a reaction. It is used for the separation and identification of a large number of compounds such as pharmaceuticals, drugs, pesticides etc.