gc/ms analysis
advertisement

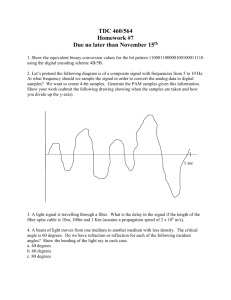
GC/MS ANALYSIS 2003 REVISION OBJECTIVE: The volatile components of a foodstuff such as wine are isolated and concentrated by headspace analysis. The components in the concentrate are separated by capillary gas chromatography and the mass spectrum of each component is measured. The mass spectra are used to identify the components. REFERENCES: Chapter 2 in R. M. Silverstein, G. C. Bassler, and T. C. Morrill, Spectrometric Identification of Organic Compounds, 5th. ed., Wiley, New York. F. W. McLafferty, Interpretation of Mass Spectra, 3rd. ed., University Science Books, Mill Valley, California, 1980. Mass Spectrometry Data Centre, Eight Peak Index of Mass Spectra, 2nd. ed., Aldermaston, U.K., 1974. W. G. Jennings, ed., Applications of Glass Capillary Gas Chromatography, Chromatography Science Series, Volume 15, M. Dekker, New York, 1981. H.-D. Belitz and W. Grosch, Food Chemistry, 2nd. ed., Springer Verlag, Berlin, 1987. "Solid Phase Microextraction: Theory and Optimization of Conditions", Technical Note T198923 (Bulletin 923), Supelco, Bellafonte, Pennsylvania, 1998. http:sigma-aldrich.com/Brands/Supelco_Home/Technical_Library/ Literature.html "Solid Phase Microextraction Troubleshooting Guide", Technical Note T101928 (Bulletin 928), Supelco, Bellafonte, Pennsylvania, 2001. http:sigma-aldrich.com/Brands/Supelco_Home/Technical_Library/ Literature.html CHOICE OF METHOD: Two methods of isolating the volatile components are now available in the experiment: headspace analysis (properly called purge-andtrap with our adaptation) and solid-phase microextraction (SPME). In the former, nitrogen is passed through or over the sample, the volatiles are trapped and dissolved in a small volume of methanol. Multiple injections are possible in this mode but the sample is diluted by the solvent. In the latter, an adsorptive fiber is 2 immersed in the sample or a slurry of the sample and the analytes are adsorbed on its surface. The fiber is injected manually in the gas chromatograph and all of the adsorbed molecules are quickly evaporated. This approach is very effective in concentrating the sample and is not limited to the most volatile components. However, a separate preparation is required for each injection. CHOICE OF SAMPLE:\ Discuss the selection of you sample with the instructor before you begin the experiment. Poor results are guaranteed with a poor selection of unknown. The experiment should work with any odiferous foodstuff. Excellent results have been obtained with vintage wines although any odiferous beverage should be amenable to attack. However, if the beverage easily forms a foam, one must use the SPME method. Solids can also be studied for headspace analysis by chopping up the material and placing the pieces in the washing bottle. In the case of SPME, one can either create an aqueous slurry with a blender and insert the fiber in the slurry or place the chopped-up pieces of the material in a closed vial and insert the fiber into the vial. Fresh mint, pineapple, orange rinds, coffee, and ripe bananas have yielded excellent results. Attempts to characterize chocolate failed. Use fresh, ripe material. Fruits lose most of their volatiles in the canning process. Bay leaves plucked off a California Mountain Laurel yield far more results than dried leaves from a store-bought bottle. METHODS OF ISOLATION OF THE VOLATILES You will chose the method of isolation in your pre-lab meeting with the instructor. I) HEADSPACE ANALYSIS: The apparatus used to isolate and concentrate the volatile components of the sample is sketched in Figure 1 at the end of this document. Nitrogen is passed through a washing bottle filled with the sample. The exit stream contains the volatile components which are trapped by a column filled with a solid support, Tenax in our case. Chromosorb 101 and 102 are also available as solid supports. After the volatiles have been collected, the column is heated and backflushed and the released material is collected in a trap cooled with liquid nitrogen. The components used in each step of the collection and the conditions are given in Table 1. 3 Table 1 Suggested Parameters for Each Step of the Headspace Analysis __________________________________________________________________ Step Washing Position Level Heater Length Trap Bottle of the of the (Variac) of the (used (used Collection Flowmeter Setting Step or not) or not) Tube(1) Ball (min.) __________________________________________________________________ 1 on A 10.0 off, 0 60 off 2 off A 7.5 off, 0 30 off 3 off B 7.5 on, 35 30 on 4 off B 7.5 on, 35 30 off __________________________________________________________________ (1) B indicates that the B end of the collection tube is connected to the nitrogen flow (the orientation shown in Fig. 1). A indicates the reverse orientation. The procedure consists of the following steps. Step 1. Fill the washing bottle with the sample. If it is a liquid, use ca. 100 ml. If it is a solid, the sample should be finely divided. The A end of the sample collection tube is connected to the outlet of the washing bottle and nitrogen is allowed to flow through the washing bottle for 60 minutes. Water and other volatile components are swept out of the bottle and trapped on the column. Step 2. The washing bottle is removed from the system and the nitrogen flow is continued for another 30 minutes. Most of the water is swept out of the collection tube during this period. The bulk of the sample should remain at the A end of the tube since it is at room temperature. Step 3. The collection tube is reversed so that the B end is connected to the outlet of the flowmeter and the U-shaped trap is connected to the A end. Purge the trap with the nitrogen flow for 2 minutes. Then cool the trap with liquid nitrogen in a small Dewar flask and turn on the heater. At the elevated temperature, the sample will be purged from the collection tube. Collect the sample for 30 minutes. After the sample has been collected, light the oxygen-natural gas torch and then disconnect the trap from the collection tube but keep it immersed in liquid nitrogen. Quickly seal off at the upper constriction in the trap with a glass torch, cover the open end of the collection tube with Parafilm, and remove the trap from the liquid nitrogen after the hot sealed-off end has cooled. Cut 4 the trap at cutting point one. This point should be above the condensate in the trap. Wash the material collected with a few drops of methanol. Invert the trap so that the solution collects at the sealed end. Cut the tube at cutting point two and transfer the solution to the mini MS sample tube which has small plastic feet. Place the sample tube in a sample vial and cap the vial with a cover with a septum. If any material remains in the collection trap, cover the open end with parafilm. N.B. You do not want to use too much methanol to dissolve the sample. Premeasure how many drops of methanol are required to almost fill the mini sample tube. Use this and no more to dissolve the condensate. Step 4. After the trap has been removed, continue the flow through the heated collection tube for another 30 minutes to insure that all material has been swept from the tube. Turn off the heater and allow the tube to cool before the system is disassembled. This procedure can be modified depending on the sample. Consider the case of analyzing mint leaves. In contrast to wine or a juicy orange, the water content is not high. Hence, one can dispense with the Tenax trap and directly collect the volatiles in the Utube cooled in liquid nitrogen. II) SOLID-PHASE MICROEXTRACTION This handout will briefly describe the use of our SPME unit which in its first use gave excellent results on coffee. Read the technical notes on the Supelco Web site for details. The working part of the SPME unit is a fragile fiber that is coated with an absorptive coating,presently 75 m of Carboxen/ polydimethylsiloxane (CAR/PDMS. When the unit is not in use or is being injected through a septum, the fiber is withdrawn into a protective needle. During the adsorption or desorption step, the fiber is extended. The following simplified procedure has worked well. A) Removal of material pre-adsorbed on the fiber. The previous user should have cleaned the fiber but one should not make this assumption. 1) Move the sample changer on the GC-MS unit to the side. 2) Load the method spme_bg.m 5 3) Turn off the flow saver and change the temperature of the GC injector from 280C to 300C. 4) Make sure that the fiber is withdrawn into the protective needle. The Adjustable Depth Gauge should be at the 3.0 position. 5) Manually inject the needle through the septum on the gas chromatogram. 6) Expose the fiber to the stream of hot helium in the injection port by pressing down on the plunger of SPME fiber holder and locking the plunger. 7) Wait 60 minutes for full desorption to occur. 8) Unlock and release the plunger on the SPME fiber holder. The fiber is now protected. 9) Remove the unit from the injection port of the GC. 10) Lower the temperature of the injector to 280C and enable the flow saver. (You will also repeat these steps at the end of the experiment.) B) Confirm that the fiber is clean with a background run. This step will be a practice run on the use of the Hewlett-Packard GC-MS. Refer to the following section that is written for a headspace analysis but note the changes to apply to SPME. In particular, you will be performing a manual rather than an automatic injection. 1) Move the automatic sample holder off to the side if you have not already done so. 2) Run the autotune procedure. You only need to do this once each day. 3 Load the method spme_bg.m. 4) Set parameters and prepare the instrument for manual injection. The instructor will review the instrumental parameters at this time. This outline will take you through a maze of menu options and mouse clicks. Method Edit Entire Method Instrument/Acquisition (Check this option.) (The other options should not be checked.) OK Sample Inlet: GC Injection Source: Manual Injection Location: Front Check the box labeled Use MS. 6 OK Real Time Plot Signal 1 Attenuation: 0 Offset: 10% Time: 5.0 min. (These are the default values.) OK MS Tune: ATUNE.U (You are selecting the file for the tuning that was defined in the autotune sequence.) OK You will now be prompted to view and change the GC settings. OK You will now be prompted to review and change the MS SIM/SCAN settings. Set the solvent delay to 0.1 minutes. There will be very little solvent to be desorbed from the fiber so a very short delay is possible. Consequently, you will not miss rapidly eluting components. OK Finally save the method. Don't change the path or the extension. The file name should not exceed 8 characters. Do not use spme_bg for the file name. 5) To execute a run, complete the following sequence of steps. a) Mouse clicks and keyboard entries. Method Run Provide a unique file name (up to 8 characters but don't change the extension D or the path.) Run Method (Here, don't click on OK.) 7 b) At first a message "Waiting for GC" will appear. The GC is being is initialized. Do not bypass this step. Be patient and wait. The message will disappear when the GC is ready. c) Wait for a second message window dealing with manual injection to appear. Press as prompted the Prep Run button on the righthand keypad of the GC. d) At first the message on the GC will indicate that it is not ready. Wait until the message changes to "STATUS Ready for Injection". e) Insert the SPME holder as instructed in step A above. Press down on the plunger and immediately press the Start button on the GC. f) You have initiated a run. After 30 seconds of exposure of the fiber to the flow of hot helium in the injection port, retract the fiber and remove the SPME holder from the GC inlet. The length of the exposure time is a parameter that you might change. g) After the GC run has been completed, examine the total ion chromatogram and the mass spectrum at selected elution times. Determine if further preconditioning of the fiber is necessary. C) Collection of adsorbables from the sample. 1) Unless necessary, we shall store the sample vial or beaker. Using a clamp, mount the SPME the beaker or vial. Adjust the height so that does not contact the sample but the fiber will plunger is depressed. in an open holder above the needle when the 2) Depress and lock the plunger. 3) Allow 15-30 minutes for adsorption to occur. 4) Retract the plunger to protect the delicate fiber. 5) Analyze the adsorbed material with the GC-MS as described in step B above. The first run should be conducted with the conventional 40:1 split. Use the first run to plan for further runs. Since a quantitative analysis is not our goal, cleaning of the fiber between runs is probably not necessary. 8 D) When you are finished with the analysis, clean the fiber as described in step A above. USE OF THE HEWLETT-PACKARD GC-MS FOR HEADSPACE ANALYSIS: The components of the concentrate are separated on a capillary glass column whose temperature is varied according to a linear program. The mass spectrometer is used as a detector for the gas chromatograph. 1) Place the sample vial prepared above in one of the slots of the automatic sample changer. 2) Start the MS software and autotune the mass spectrometer. Select the standard spectra feature that optimizes the spectrometer for generating spectra useful in library searches. 3) Define the method. That is, set up the instrument for a run. Normally the protocol is defined so that no single component exceeds 250 ng. Consider the following example which will be assumed to apply in our case. Suppose that a 1 wgt-% solution of an unknown in methanol is prepared. A 1 l sample of the solution (this is a typical injection volume) would contain 10 g of the unknown, a value far in excess of the 250 ng. A split ration of at least 40 is required. That is, only 1/40’th of the outlet gas stream from the gas chromatograph is passed on to the mass spectrometer and the remainder is rejected. Use a split ratio of 40 for the first run and decrease it if the signal-to-noise ratio is poor. a) Load the c160_vin default method. Here are some of the features of the method. The instructor will review all of the parameters during the prelab orientation. i) helium flow rate: 1 ml/min ii) initial column temperature: 70C iii) time at 70C: 2 min. iv) ramp rate for the column: 10/min. v) final column temperature: 200C vi) total length of the run: 20 min. vii) mass range: 40-600 amu viii) split ratio: 40:1 ix) selective ion monitoring at 88 (This is appropriate for the analysis of wines since the Maclafferty 9 rearrangement yields a strong peak here for ethyl esters of straight-chain aliphatic acids.) x) solvent delay: 1.5 min. (This number is very important as the high voltages in the mass spectrometer are not turned on until after the solvent peak has eluted from the column. The number is is determined manually by measuring the time between injection and a rise in the forepump pressure. The value given above is for methanol under the specified operating conditions and must be checked for each solvent and set of operating conditions.) b) Modify the method as appropriate. c) Save the method under a unique name that you select. Do not change the path or extension. d) Run the method. Provide a unique file name for the results and the slot number for the vial. e) Analyze the results and conduct additional experiments. There are a number of scenarios. i) None of the peaks are strong. Conduct another run with a lower split ratio. If the peaks are very weak, consider running splitless, i.e. all the GC effluent enters the MS. ii) The strong peaks elute early and are followed by weak peaks. Consider an additional splitless run BUT increase the solvent delay time to slightly longer than the last strong peak. iii) The strong peaks elute in the middle of the run and are preceded and followed by weak peaks. Two additional runs are required. For one, apply the strategy just discussed. Run splitless but increase the solvent delay to exclude the strong peaks. This run will better characterize the weak but slowly eluting components. To improve the sensitivity of the weak components that elute early, run splitless but design the schedule so that the mass spectrometer is shut down before the strong peaks elute. A post run (i.e. the GC component of the run continues even though the MS is turned off) is required to elute all the components from the GC column before the next run. 3) Analyze the results. Average the mass spectrum of each GC peak over the center of the peak and apply background correction. If you suspect that the peak has two components, compare the mass spectrum at the front and the rear of the peak. What would happen if two substances elute with slightly different elution times only one peak is resolved? Perform a library search and identify the 10 components. Our computer has a subset of the NIST MS database. Use a visual comparison to assist in making your decision. Chemical common sense has its place. Would tetraethylsilane or atropine be likely components of wine? REPORT: Your report should contain the following components: 1) a list of all identified components, their relative amounts, and the basis for the identification. Relate the components to the known chemistry of the material analyzed. For example, orange rinds contain terpenes and the isoprene rule applies. The excellent NIST Web page that is cited by MolData provides links to the full NIST MS database. The NIST link is found on the Physical Chemistry page. The SDBS Japanese database cited on the Organic Chemistry page of MolData is superb. The URL for MolData if you have forgotten is pages.pomona.edu/~wsteinmetz/moldata.html. 2) an assignment of the principal peaks of the 4 most abundant components. If you have 4 or less components, present an assignment of all components. The cited works on mass spectrometry will be helpful in this step. A sample assignment is provided with this handout. 3) a comparison of your analysis with what others have observed. The excellent monograph by Belitz and Grosch is an excellent place to start. Some use of the primary chemical literature is expected. gcms.doc, WES, 9 Jan. 2003