MIAPE_Quant_v1.0_ICPL_VH
advertisement

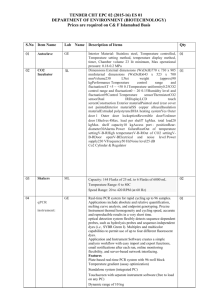
Classification 1. 2. Definition General features — Global descriptors of the experiment 1.1. Experiment identifier or name Regulation of proteins in AIRE-overexpressing cells 1.2. Responsible person or role Joan Josep Bech / Francesc Canals (Laboratori de Proteòmica-VHIO). Tef.934894174. 1.3. Quantitative approach ICPL Experimental design and sample description —2.1 Experimental design 2.1.1 Groups Group 1: AIRE-overexpressing cells. Group 2: Control cells. 2.1.2 Biological and technical replicates Group 1: AIRE-overexpressing cells. Group 2: Control cells. 2. Experimental design and sample description —2.2 Sample / Assay description Labelling protocol (if applicable) Standard ICPL labelling protocol from the manufacturer at protein level. 2.2.2 Sample description Sample name AIRE: AIRE-overexpressing cells. CTRL: Control cells. 2.2.2.2 Sample amount 100 micrograms of a mixture of both heavy- and light-labeled samples were loaded into the 1D-gel before the first fractionation step (see section 3.3). 2.2.2.3 Sample labelling with assay definition, i.e. MS run / data set together with reporting ion mass, reagent or isotope labelled amino acid ICPL-12C: 105.0215 Da ICPL-13C: 111.0419 Da m: 6.0204 Da 2.2.2.3 Replicates and/or groups ICPL2: AIRE (ICPL-12C) and CTRL (ICPL-13C). ICPL1: AIRE (ICPL-13C) and CTRL (ICPL-12C). 2.2.3 Isotopic correction coefficients N/A 2.2.4 Internal references N/A 3. 4. Input data — Description and reference of the dataset used for quantitative analysis: type, format and availability of the data. No actual values are requested here. 3.1. Input data type EICs from full MS scan 3.2. Input data format .yep files (bruker) 3.3. Input data merging Prior to the analysis by mass spectrometry, both AIRE and CTRL samples were fractionated in 20 bands by using a 1D-gel. Each individual band was subsequently digested and submitted to a second chromatography-based fractionation step before the analysis into the mass spectrometer. Finally, the searches for the 20 individual bands were merged in a unique compilation list by using ProteinScape 2.0 software, before the quantification analysis. 3.4. Availability of the input data URI Protocol —Description of the software and methods applied in the quantitative analysis (including transformation functions, aggregation functions and statistical calculations). 4.1. Quantification software name, version and manufacturer WARP-LC 1.2 / ProteinScape 2.0 (Bruker) 4.2. Description of the selection and/or matching method of features, together with the Features (EICs) were selected by applying a m/z and retention time window of 1.3Da and 60 sec, respectively. Only signals from +2 and +3 species were added to each peptide EIC. The values used for quantitation were obtained by description of the method of the primary extracted quantification values determination for each feature and/or peptide integrating the chromatographic peak of each EIC. 4.3. Confidence filter of features or peptides prior to quantification A FDR <4% is applied (Described in MIAPE-MSI xx). 4.4. Description of data calculation and transformation methods 4.4.1. Missing values imputation and outliers removal Outliers are automatically removed by WARP-LC 1.2 software. 4.4.2. Quantification values calculation and / or ratio determination from the The heavy/light ratios (H/L) of each peptide were calculated by dividing the intensities of the EICs corresponding to the heavy (ICPL-13C) labeled- and light (ICPL-12C) labelled- versions of each peptide. primary extracted quantification values 4.4.3. Replicate aggregation N/A 4.4.4. Normalization The normalization is performed by using the protein quantification, which is calculated as indicated in section 4.4.5. Then the distribution of frecuencies of protein ratios were calculated. Finally, The Mode value of the distribution is used to re-center the distribution to 1. 4.4.5. Protein quantification values calculation and / or ratio determination from the peptide quantification values 4.4.6. 5. Protocol specific corrections H/L ratios for proteins were calculated by performing the arithmetic mean of all the ratios obtained at peptide level after outlier removal. N/A 4.5. Description of methods for (statistical) estimation of correctness A coefficient of variation (CV) was calculated and only proteins with CV<30% were accepted. 4.6. Calibration curves of standards N/A Resulting data —Provide the actual quantification values resulting from your quantification software together with their estimated confidence. Depending of the quantification technique or even of the quantification software, only some of the following items could be satisfied (e.g., for spectral counting, only quantification values at protein level can be provided) 5.1 Quantification values at peptide and/or feature level: Actual quantification values achieved for each peptide and/or, in case of feature-based quantification, for the corresponding features (mapped back from each peptide), together with their estimated confidence. 5.1.1. Primary extracted quantification values for each feature (e.g. area, height, etc.), with their statistical estimation of correctness 5.1.2. Quantification values for each peptide as a result of the aggregation of the values of the previous section (5.1.1), with their statistical estimation of correctness N/A http://proteo.cnb.csic.es/downloads/miape-quant/Peptides_ICPL1_VH.xls http://proteo.cnb.csic.es/downloads/miape-quant/Peptides_ICPL2_VH.xls 5.2 Quantification values at protein level: Actual quantification values achieved for each protein and for each protein ambiguity group, together with the confidence in the quantification value. Basic / raw quantification values with statistical estimation of correctness N/A 5.2.2. Transformed / aggregated / combined quantification values of the proteins at group level, with their statistical estimation of correctness http://proteo.cnb.csic.es/downloads/miape-quant/Proteins_ICPL1_VH.xls http://proteo.cnb.csic.es/downloads/miape-quant/Proteins_ICPL2_VH.xls