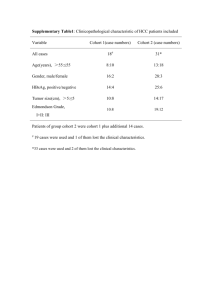
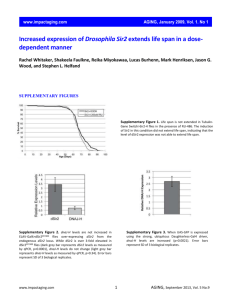
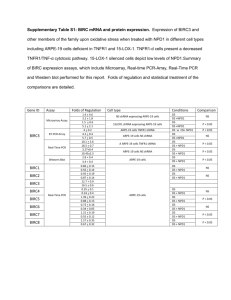
Supplementary Methods
advertisement

Molloy-Simard et al., supplementary material 1 SUPPLEMENTARY MATERIAL Supplementary Methods Construction of recombinant plasmids pCAT-Basic-HuPARG was linearized with both SphI and PpMUI (NEB CANADA, Toronto, ON, CANADA) and then digested with 20 µl ExoIII (NEB CANADA, Toronto, ON, CANADA) at 35°C. Aliquots were taken every 10 sec and inactivated for 10 min at 70°C. Afterwards, 0.5 µl of Mung bean nuclease (NEB CANADA, Toronto, ON, CANADA) was added to each aliquot and further incubated for 1 h at 30°C. The DNA segments were precipitated, resuspended in water and the size of the digested plasmids verified on agarose gel [0.8% agarose (m/v), 0.5 µg ethidium bromide]. 1 µl T4 polymerase (NEB CANADA, Toronto, ON, CANADA) and 1 µl of dNTPs 10 mM (Invitrogen) were added to the DNA at 37°C for 1 h. DNA was finally extracted with phenol/chloroform and precipitated, religated with T4 DNA ligase (NEB CANADA, Toronto, ON, CANADA), transformed in DH5α cells, sequenced and kept at -20°C. Derivatives from the plasmid huPARG(-160) that bear mutations into either the -76 or the -53 ETS target site, or into both ETS sites were constructed following the supplier’s recommendations. Briefly, 18 cycles of amplification with a five-minutes extension were used to generate the mutations on both target sites (ETS-76 and ETS-53). The recombinant plasmid pCAT-Basic-HuPARG(-160) was used as template and the primers are described in Supplementary Table 1. Primer Muta-ETS-76F and MutaETS-76R were used to change the sequence 5’-TTCCGCTTTG-3’ from the -76 ETS site into 5’TTTTGCTTTG-3’ while Muta-ETS-53F and Muta-ETS-53R were used to change the sequence 5’CCGGAAGCT-3’ from the -53 ETS site into 5’-CCAAAAGCT-3’. The resulting recombinant plasmids were named Hu-PARG(-160/mETS-76) and Hu-PARG(-160/mETS-53), respectively. The latter was used as a template to generate the plasmid Hu-PARG(-160/mETS-76:-53) in which both ETS Molloy-Simard et al., supplementary material 2 target sites were mutated. The Splicing by Overlap Extension method (SOE) (1) was used to create a deletion of the entire region between both target sites in pCAT-Basic-huPARG(-160). The promoter sequence upstream of ETS-76 was fused with the sequence downstream of ETS-53, creating the needed deletion in between (-79 to -53 inclusively). Briefly, the upstream portion was amplified by standard PCR using primers Del-ETS-Ext-F and Del-ETS-Int-R while the downstream portion was amplified using primers Del-ETS-Int-F and Del-ETS-Ext-R. The internal primers Del-ETS-Int-R and Del-ETSInt-F were design so that their 5’ ends are complementary to the 3’ end of one another. These portions of the promoter were then purified and fused together using ten cycles of amplification with a 25minutes annealing at 50ºC and a five-minutes extension at 72ºC. The resulting fusion was then purified and used as template for a final PCR amplification using the external primers Del-ETS-Ext-F and DelETS-Ext-R. These two primers are flanked with the restriction sites SalI and XbaI respectively, which enable cloning of this fragment back into the vector pCAT-Basic. The resulting recombinant plasmid was named huPARG(ΔETS-79/-53) and transfected into uveal melanoma cell lines. Lentivirus production Target sequences for the production of shRNAs against the human PARG mRNA transcript were selected using BLOCK-iT RNAi Designer (Invitrogen). The same process was used to select the shRNAs directed against the human PARP-1 transcript except that one of the three sequences was selected according to Shah et al. (2) and corresponded to the SiP912 target sequence cited. This specific target sequence was reported to yield an shRNA very efficient at knocking-down PARP-1 expression. Three different target sequences were selected for each gene and oligonucleotides were designed with flanking sequences on both sides (the DNA sequence of those that proved to be the most effective at suppressing expression of PARG and PARP-1 are shown on the Supplementary Table 4). BglII and HindIII restriction sites were added on the 5’-end and 3’-end respectively along with a Molloy-Simard et al., supplementary material 3 CGAA loop in between. The sense and anti-sense oligonucleotides were fused together and cloned into the pSuper vector using BglII and HindIII. To extract the fragment containing the H1 promoter fused to the shRNA sequence, pSuper-shRNA was then cut with ClaI and XbaI. This fragment was then cloned into the lentivirus vector pNL-SIN-GFP (kindly provided by Dr. Bryan R. Cullen, Duke University Medical Center, Durham, N.C., USA). The resulting recombinant plasmid contains the H1 promoter to drive the expression of the shRNA and the CMV promoter to drive the expression of the GFP. Another plasmid was made to target the luciferase gene as negative control. For the production of virus, 8x106 cells were plated onto 175cm2 tissue-culture flask and allowed to grow overnight. The medium was refreshed three hours before the cells were transfected using the calcium phosphate precipitation method (3). The DNA mixture transfected consisted of 20 µg of pNLSIN-GFP-shRNA, 1.1 µg of pcRev, 1.1 µg of pcTat and 0.55 µg of pVSV-G. The medium was replaced 18 h later with fresh medium containing 2.5 % FBS. Two days later, the medium was harvested and centrifuged at 3 000 g for 10 min to remove debris. The medium was then centrifuged at 50 000 g for 2 h at 4°C to collect viruses. Viruses collected were resuspended in 500 µl of medium containing 10 % FBS and stored at -80°C. Molloy-Simard et al., supplementary material 4 Supplementary references 1. 2. 3. Horton, R. M., Hunt, H. D., Ho, S. N., Pullen, J. K., and Pease, L. R. (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension, Gene 77, 6168. Shah, R. G., Ghodgaonkar, M. M., Affar el, B., and Shah, G. M. (2005) DNA vector-based RNAi approach for stable depletion of poly(ADP-ribose) polymerase-1, Biochem Biophys Res Commun 331, 167-174. Pothier, F., Ouellet, M., Julien, J. P., and Guerin, S. L. (1992) An improved CAT assay for promoter analysis in either transgenic mice or tissue culture cells, DNA and cell biology 11, 8390.