PowerPoint Presentation from June
advertisement

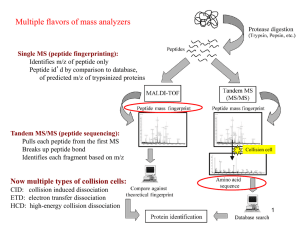
Protein Identification and Peptide Sequencing by Liquid Chromatography – Mass Spectrometry Detlef Schumann, PhD Director, Proteomics Laboratory Department of Genome Science May 27, 2005 The Proteomics Problem Cell state 1 Protein 1 - Protein name: ... - MW: ... - Amino acid sequence: ... - Modifications: ... Why are state 1 and 2 different? Cell state 2 Proteomics Protein 2 -Protein name: ... - MW: ... - Amino acid sequence: ... - Modifications: ... • • • The Typical Proteomics Problem Sample #488 Sample #487 MW 200 10 4 7 pI 4 7 pI The Proteomics Laboratory at the GRI Electrophoresis Laboratory • 1-D gel electrophoresis (small format) • 2-D gel electrophoresis (small and large format) • Silver staining and Coomassie staining • Imaging densitometry of protein gels • Comparative 2-D gel data analysis • Western blotting (small format gels) • HPLC separation of protein mixtures Mass Spectrometry Laboratory • Peptide mass fingerprinting • LC-MS/MS analysis • Analysis of protein modifications • Purity analysis of recombinant proteins/synthetic peptides • Purity analysis of oligonucleotides Basics of Protein Mass Spectrometry • Mass spectrometry determines the molecular weight of chemical compounds by separating molecular ions in a vacuum according to their mass-to-charge ratio (m/z) • Ions are generated by induction of either the loss or the gain of a charge (protonation, deprotonation or electron injection) • Generated ions can be fragmented in the vacuum, and the resulting subfragments can provide information about the structure of a compound Ion source Mass analyzer Detector Ion generation Ion separation Ion detection F. Lottspeich and H. Zorbas, Bioanalytik 1998, Spektrum Akad. Verlag Mass Spectrometry Instrumentation at the GRI 1. Bruker Biflex III MALDI-TOF mass spectrometer • mid fmole protein/peptide analysis • protein identification using peptide mass fingerprinting • oligonucleotide mass/purity analysis • biomarker analysis 2. Finnigan LCQ Deca XP Max ESI mass spectrometer • coupled to Dionex Ultimate nanoflow 2-D HPLC • low fmole peptide analysis • protein identification using LC-MS/MS peptide sequencing 3. PE Sciex API 3000 ESI mass spectrometer • low pmole/high fmole peptide/metabolite analysis • identification of post-translational modifications • peptide and metabolite quantitation studies Protein Identification by Mass Spectrometry 1. Peptide Mass Fingerprinting • protease digestion of protein spots/bands • peptide extraction • sample spotting on target plate • mass measurement of peptide ions by MALDI-TOF MS or LC-MS • data base search using generated mass list • protein identification based on ≥ 4 matched peptide masses 2. Peptide Sequencing • protease digestion of protein spots/bands • peptide extraction • RP-LC separation of peptides • mass measurement and fragmentation analysis of peptide ions • data base search using parent mass and fragment mass data • protein identification based on ≥ 2 matched peptides Intens. [a.u.] Peptide Mass Fingerprinting x10 4 1091.620 Sample: in-gel digested human EF-2 1.5 1799.879 1347.669 1.0 2143.156 1615.722 890.612 1214.658 0.5 1504.667 1978.039 2460.281 2801.340 0.0 1000 1500 2000 2500 3000 m /z Peptide Mass Fingerprinting Result Peptide Mass Fingerprinting Result Tandem Mass Spectrometry (MS/MS) Analysis eluting peptide mass analysis precursor ion fragmentation fragment mass analysis Tandem Mass Spectrometry (MS/MS) Analysis eluting peptide mass analysis precursor ion T E S T P E P T I D E+ b-ions b1 b2 b3 b4 b5 b6 b7 b8 b9 b10 b11 T+ TE+ TES+ TEST+ TESTP+ TESTPE+ TESTPEP+ TESTPEPT+ TESTPEPTI+ TESTPEPTID+ TESTPEPTIDE+ - H2O fragmentation fragment mass analysis T E S T + P E P T I D E+ TESTPEPTIDE+ ESTPEPTIDE+ STPEPTIDE+ TPEPTIDE+ PEPTIDE+ EPTIDE+ PTIDE+ TIDE+ IDE+ DE+ E+ y11 y10 y9 y8 y7 y6 y5 y4 y3 y2 y1 y-ions LC-MS/MS Analysis of Protein Digests NL: 100 1.21E8 100 Relative Abundance Relative Abundance 80 60 40 Base Peak Y12 1299 NL 5.29E6 Base peak Y6 689 B6/Y132+ 706 Y4 475 Y122+ 650 B4 493 B3 380 Y10 1087 Y7 803 Y5 588 Y8 902 B9 990 B10 1102 B11 1204 B12 1317 Y13 1412 Y11 1202 0 400 800 1200 1600 m/z 20 0 10 15 20 25 30 35 40 45 50 55 60 Time (min) Base peak chromatogram of the LC-MS/MS analysis of a protein digest from a silver stained 2D gel spot, the insert showing the MS/MS spectrum for the actin peptide SYELPDGQVITIGNER as identified by SEQUEST LC-MS/MS Analysis of Protein Digests I/L Relative Abundance 100 T I/L V QG D P I/L Y12 1299 NL 5.29E6 Base peak Y6 689 Y4 475 Y10 1087 Y122+ 650 B4 493 B3 380 B10 1102 B6/Y132+ 706 Y7 803 Y5 588 Y8 902 B9 990 B11 1204 B12 1317 Y11 1202 Y13 1412 0 400 800 1200 m/z Peptide sequence: SYELPDGQVITIGNER 1600 LC-MS/MS Analysis Result LC-MS/MS Analysis Result Frequently Asked Questions 1. How much protein do you need? Short Answer: At least 1 pmol Factors: Long Answer: It depends ... - protein staining - protein sequence - protein size - potential post-translational modifications - presence of the protein sequence in the database 2. When can I get the results? Short Answer: In 1-2 weeks Factors: Long Answer: It depends ... - type of requested analysis - amount of protein sample - protein sequence - protein size - potential post-translational modifications - presence of the protein sequence in the database Frequently Asked Questions 3. I saw a dark band/spot on the gel. Why did we get no results? 1 2 3 4 5 6 7 8 Loading (100 ng protein/lane): 1+2 Ovalbumin (Chicken) 3+4 Myoglobin (Horse) 5+6 Cytochrome C (Horse) 7+8 Serum albumin (Bovine) Ovalbumin ~ 45 kDa Myoglobin ~ 17 kDa Cytochrome C ~ 13 kDa Serum albumin ~ 66 kDa 100 ng ~ 2.2 pmol 100 ng ~ 5.9 pmol 100 ng ~ 7.9 pmol 100 ng ~ 1.5 pmol The Limitations 1. Protein Size Small proteins ( 10 kDa) or large proteins ( 150 kDa) are more challenging to digest and analyze because they generate few peptides (small proteins) or show increased resistance to proteases (large proteins). 2. Protein Sequence Proteins are typically digested using trypsin (K/R cleavage); the distribution of these AA dictates the size and the detectability of the generated peptides. 3. Post-translational Modifications Glycosylated proteins show high resistance to proteases; certain post-translational modifications (e.g. phosphorylation) decrease the detectability of the modified peptide using the standard protein mass spectrometry techniques. 4. Protein Sequence Databases The database search algorithms compare the generated spectra with theoretical digests of proteins in protein sequence databases; the positive identification of the analyzed protein depends on the presence of its sequence in those databases. The Big No-No’s 1. Detergents Detergents used for extraction and purification of proteins, when not completely removed, can cause signal suppression and decreased detectability of peptides in the mass spectrometry analysis 2. Contaminants In-gel digests of low abundance samples are very sensitive to the presence of contaminants, particularly contaminating proteins. The handling of samples/gels with gloves is absolutely necessary and the use of designated equipment for specific separation and staining protocols is highly recommended. 3. Formaldehyde or Glutaraldehyde Fixation in Silver Staining While increasing the staining sensitivity, these fixation steps result in a covalent modification and cross-linking of proteins, which can result in decreased digestion efficiency. Contact Information Laboratory Address Proteomics Laboratory Department of Genome Science (ML 0505) Genome Research Institute University of Cincinnati Building B, Room 131 2180 East Galbraith Road Cincinnati, Ohio 45237 Tel: 513/558-8950 Fax: 513/558-5061 Email: detlef.schumann@uc.edu Staff Members • Detlef Schumann • Wendy Dominick • Michael Wyder • Margaret Minges