Familial Hypercholesterolemia: Background Information
advertisement

Familial Hypercholesterolemia: Background Information
James A. Underberg, MD, MS, FACPM, FACP, FASH, FNLA
Clinical Assistant Professor of Medicine NYU School of Medicine
NYU Langone Center for Cardiovascular Disease Prevention
Director, Bellevue Hospital Lipid Clinic, New York,NY
Specialist
Disclosures
• Honoraria for Speakers Bureau (Pharma) : AstraZeneca, Abbott,
Forest, GlaxoSmithKline, Daiichi-Sankyo, Kowa, Novartis, Pfizer,
Liposcience, diaDexus, Merck, Eli Lilly
• Honoraria for CME Programs :American Heart Association,
National Lipid Association, American College of Reproductive
Medicine, PriMed, Primary Care Network
• Consulting Income: Liposcience, Amarin, Genzyme,News
Corporation, Publicis Inc., Summer Street Consulting Inc.
Guidepoint Global
• Advisory Boards: Kowa, Abbott, Merck, Genzyme, Amarin
• Clinical Research Funding: Genzyme, GlaxoSmithKline, Kowa
• Medical Education Committee Member : ASH, NLA
• Editorial Board Member: Journal of Clinical Lipidology
• Scientific Advisory Board: FH Foundation
• Board of Directors: NLA, Foundation of the NLA, ASH Foundation
FH: A Clinically Recognizable Genetic Disorder
• Inheritable, autosomal dominant disorder1
• Usually due to mutations in LDL receptor gene2,3 that result in
decreased clearance of LDL particles from plasma1
– Other mutations include those in the Apo B and PCSK9
genes
• Clinical manifestations include1,2
– Severe hypercholesterolemia due to accumulation of
plasma LDL
– May be accompanied by cholesterol deposition in tendons
and skin (xanthomas) and in the eyes
– Evidence of CVD early in life
1. Marais AD. Clin Biochem Rev. 2004;25:49-68.
2. Mahley RW, et al. In: Kronenberg: Williams Textbook of Endocrinology. 2008.
3. Rader DJ, et al. J Clin Invest. 2003;111:1795-1803.
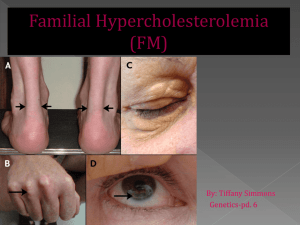
Visible Signs of FH
A- Xanthelasma
B – Corneal arcus (Arcus senilis)
C - Achilles tendon xanthomas
D - Tendon xanthomas
E - Tuberous xanthomas
F - Palmar xanthomas
Mahley RW et al. In Kronenberg: Williams Textbook of Endocrinology 2008
Genetics
• Mutations in a gene on one of the first 22 non-sex
chromosomes can cause autosomal disorders
• Autosomal Dominant
– Only one copy of the abnormal gene is adequate to cause
the disorder
– The abnormal gene dominates the pair of genes
– A child has 50% of chance of inheriting the disorder even if
only one parent has the dominant gene
• Autosomal Recessive
– Two copies of an abnormal gene must be present to cause
the disorder
– People with only one defective gene are considered
carriers
– A child has a 25% chance of inheriting the disorder if both
parents carry an autosomal recessive mutation
The Phenotype of FH May Reflect LDL-R, Apo B,
or PCSK9 Mutations
• LDLR codes for the LDL Receptor, which clears LDL particles from the
circulation by binding to surface Apo B
• PCSK9 induces degradation of LDLR
• FH may be caused by mutations in Apo B, LDL-R, or PCSK9
Apo B (site where receptor binds
to LDL particle)
Extracellular Fluid
LDL Particle:
Cell membrane
PCSK9
Cytosol
LDL receptor
1. Kumar: Robbins and Cotran. Pathologic Basis of Disease, 2009.
2. Rader DJ et al. J Clin Invest. 2003;111:1795-1803
Image reproduced from: http://www.dls.ym.edu.tw/ol_biology2/ultranet/Endocytosis.html.
FH Is Not a Rare Genetic Disease:
Prevalence is 2x Other Inherited Conditions
Frequency per 1,000 Births of
Common Genetic Disorders1
2.0
2
FH
1Familial
2Sickle
Neurofibromatosis
combined hyperlipidemia has a frequency of 1:200 births; however, the genetic cause is unknown.
cell disease varies greatly by ethnicity.
1. Genetic Alliance UK. Available at http://www.geneticalliance.org.uk/education3.htm.
2. Streetly A, et al. J Clin Path. 2010;63:626-629.
Prevalence Is Much Higher in Specific
Sub-populations or “Founder Groups”
North America and Europe:
HeFH ~1:500 HoFH ~<1:106
Higher incidence of HoFH:
Québec, Tunisia, South Africa, Lebanon
Naoumova RP, et al. Curr Opin Lipidol. 2004;15:413-422.
In Founder Groups, FH Prevalence Can
Be 8x Greater vs. General Population
Comparison of FH Prevalence Rates Across
Populations
1:100 to
1:72
1:500
1:270
HeFH
(US &
Europe)
Austin MA, et al. Am J Epidemiol. 2004;160:407–420.
1:170
1:165
1:67
Patients With FH Are at Very High CVD Risk Before Age 40,
Relative to the General Population
Risk of CHD in FH patients / risk of CHD in general
population
Men (n = 605)
*
Women (n = 580)
*
*
*
Scientific Steering Committee. Atherosclerosis. 1999;142:105-112.
* P <0.01 vs general
population.
*
*
*
Despite the Importance of Early Detection,
FH Is Under-diagnosed (US)
Percentage of patients
• The WHO estimated in 1999 that <10% of US patients with FH
were diagnosed
Percentage of FH patients diagnosed
<
<
World Health Organization Human Genetics Program. http://whqlibdoc.who.int/hq/1999/WHO_HGN_FH_CONS_99.2.pdf.
MI Rates in FH patients vs. Non-Statin Rx and
Normals
Versmissen J, et al. BMJ. 2008;337:a2423.
Reduction in Mortality in Subjects With Homozygous
Familial Hypercholesterolemia Associated With Advances in LipidLowering Therapy
Circulation. 2011;124:2202-2207
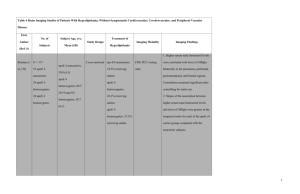
FH “Scoring Methods” for Clinical Diagnosis Require LDL Levels
and Family History
Comparison of FH Clinical Diagnostic Criteria by Method
Simon Broome Register1
Definite FH
• TC or LDL levels
• Tendon xanthoma in
patient or relative
MEDPED2
• TC or LDL levels based on
family history and age (eg,
age <20 y, with an FH
relative)
Probable FH
• TC or LDL levels
• Family history of early
MI or high TC/LDL
1. As summarized in: Marks D, et al. Atherosclerosis. 2003;168:1-14.
2. As summarized in: Civiera F, et al. Circulation. 2004;173:55-68.
Dutch Lipid Clinic
Network1
• Score based on :
Family history of
premature CHD, high
LDL, or xanthoma
Clinical history of
premature CAD or
vascular disease
Presence of xanthoma
or arcus cornealis
LDL panel
MEDPED criteria for the diagnosis of familial hypercholesterolemia. Total
and LDL cholesterol (mmol/l)band {mg/dl} criteria for diagnosing
probable heterozygous familial hypercholesterolemia
Williams RR, Hunt SC, Schumacher C, et al. Am J Cardiol 1993; 72:171–176.
Curr Opin Lipidol 2012, 23:282–289
Simon Broome Criteria
Curr Opin Lipidol 2012, 23:282–289
Dutch lipid clinic network criteria for familial hypercholesterolemia
Curr Opin Lipidol 2012, 23:282–289
World Health Organization. Familial hypercholesterolaemia. Report of a
second WHO consultation. Geneva: World Health Organization; 1999.
Role for Genetic testing in screening varies worldwide
• Testing used as a significant part of algorithms for
screening and diagnosis in many countries such as
Spain, Wales, the Netherlands, UK (NICE Guidelines)
but not currently in the US
• Use differs from country to country
• One study done in the Netherlands suggests that
with extensive screening the proportion of those
with a genetic mutation is unknown may be as low as
5%.
Curr Opin Lipidol 2012, 23:282–289
van der Graaf A, Avis HJ, Kusters DM, et al. Circulation 2011; 123:1167–1173.
Screening Varies From Country to Country
• US : NLA Recommendations (2011)Index case identified from
one of three available diagnostic criteria with universal
screening, then cascade screening of relatives in primary care
setting- genetic testing not recommended routinely
• UK: NICE guidelines (2008) Index case identified clinically
using Simon Broome followed by genetic testing and then
cascade targeted genetic screening of relatives .
• Netherlands: DLCN identification followed by genetic testing.
If mutation identified, registry in Foundation for Detection of
Hereditary Hypercholesterolemia. Then first degree family
members are genetically screened by home health nurses
followed by other family member testing.
Aarden E, Van Hoyweghen I, Horstman K. Scand J Public Health 2011; 39:634–639.
DeMott K, Nherera L, Shaw EJ, et al. London: National Collaborating
Centre for Primary Care and Royal College of General Practitioners; 2008.
Goldberg AC, Hopkins PN, Toth PP, et al. J Clin Lipidol 2011; 5:133–140.
National Collaborating Centre for Primary Care (UK). (2008).
NICE clinical guideline 71: Identification and management of
familial hypercholesterolaemia, London
Family Screening Has Dramatically Increased
Treatment Rates in the Netherlands
Effects of Family-Based Screening on Treatment
Rates in People with FH
N = 5,442
• 37% identified as HeFH (based on LDL-R mutations)
Umans-Eckenhausen MAW, et al. Lancet. 2001;357:165–168.
Role of Genetic Typing in FH
Highlights from this discussion include the role of genetic
typing for diagnosis
Understanding disease mechanism
Potential guidance in treatment algorithms
Journal of Clinical Lipidology, Vol 6, No 3, June 2012
Highlights
• “Some studies have suggested that the individuals with gainof-function mutations in PKSK9 have greater levels of LDL-C,
and although they are decreased with statin therapy, they
remain greater than in patients with low-density lipoprotein
receptor (LDLR) mutations.”
• “Today, this might influence expectations of therapeutic
effectiveness, but tomorrow might indicate which class of
cholesterol lowering drugs might be most effective.”
• Potential role for increasing treatment rates in children with
mutations identified in parents with FH
– “big benefit is for the family of someone with a known
mutation.”
CASCADE SCREENING
“The clinical validity and utility of cascade
screening for FH is dependent on a number of
factors, including the criterion used to diagnose
the disorder in the index case, the use of DNA
testing in the index case and in relatives, and the
nature of the benefit and possible harms of
identifying and pharmacologically treating the
disorder in childhood. Nevertheless, cascade
screening is a straightforward and highly
effective way to identify persons who have FH.”
Ned, R. M., & Sijbrands, E. J. (2011). Cascade screening for familial hypercholesterolemia (FH).
PLoS Curr., 3 doi:10.1371/currents.RRN1238
Screening of Children
• 2008 American Academy of Pediatrics- Family history of
premature CVD screen at age 2
• 2011 NLA- Screen all children age 9-11, and at age 2 if
family history of premature CVD
• 2012 NHLBI- screen all children between ages 9-11 and
again between ages 17-21 with earlier screening in high
risk children
• Recommendations have generated controversy- long
term effects, no hard outcome studies, anxiety, missed
diagnosis
• Australia- Universal Screening not recommended, screen
those with family history or as part of cascade testing
• UK, Netherlands, Norway- Children screened as part of
cascade testing, not universally .
Curr Opin Lipidol 2012, 23:282–289
Summary
• Heterozygous FH is a common disorder associated with
a significantly increased risk of CVD
• Observational data suggests those treated with statins
have reduce risk to unaffected levels
• Disease is underdiagnosed
• Screening promotes treatment
• Screening in US is based on clinical criteria with no
current recommendations for routine genetic testing
• Role for genetic testing varies internationally, and may
increase with reductions in cost