This Lecture....
advertisement

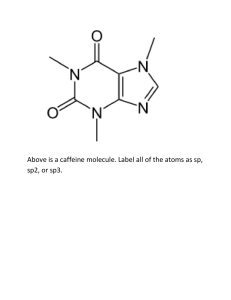
This Lecture.... Is born from the ever growing realisation that the drug discovery turn-over rate must be rendered more efficient... Both from the perspective of innovator companies & from that of the ultimate beneficiary of the process i.e. the patient Chronologically….. Chris Lipinski: Drug-like is defined as those compounds that have acceptable ADME/tox properties to survive through the completion of human Phase 1 trials And… Ronald T. Borchardt: Drug-like responses are intrinsic properties of the molecules, and it is the responsibility of medicinal chemists to optimise not only the pharmacological properties, but also the drug-like properties of the molecules From the Previous Quotations it is Possible to Infer….. The term drug-like with respect to a drug molecule implies that certain properties of a particular compound confer on that molecule a greater propensity to become a successful drug product The Major Pioneer in This Field.. Is Chris Lipinski. Examined the structural properties that affect the physico-chemical properties of solubility and permeability, and their effect on drug absorption Puts forward notion that many properties are of interest in drug discovery…. The Structural Properties of the Molecule Hydrogen Bond Forming Moieties Lipophilicity Molecular Weight Polar Surface Area Shape Reactivity pKa The Physico-Chemical Properties Solubility Permeability Chemical Stability The Biochemical Properties Metabolism (Phases 1 & 2) Protein and Tissue Binding Transport Modality Pharmacokinetics & Toxicity Clearance Half-Life Bioavailability Drug-Drug Interactions Structure Determines a Compound’s Properties Pharmacokinetics & Toxicity Clearance, Halflife, Bioavailability, LD50 Physicochemical Properties: Solubility, Permeability & Chemical Stability Biochemical Properties: Metabolism, Transporter Affinity, Binding Target Affinity Physical Environment Structural Properties: Molecular Weight, Hydrogen Bonds, Lipophilicity, PSA, pKa, Shape, Reactivity Proteins Therefore it May Be Inferred That… Interaction between structural properties and the physical environment characterises the physicochemical properties of a molecule eg solubility Interaction between structural properties and proteins characterises the biochemical properties of a molecule eg metabolism At the highest level, the interaction between physicochemical & biochemical properties & living systems characterises the pK & toxicity of a molecule The Corollary Therefore is …. That it is possible for medicinal chemists to control the pK and toxicity properties of a molecule through the modification of its structure The Drug Discovery & Development Process: DISCOVERY •Biological Target •ID & Characteristics •Activity & Selectivity •Chemical Synthesis •Property Profiling DEVELOPMENT •Batch synthesis •Analytical Release •Formulation & stability •Human Efficacy •Safety & pK CLINICAL APPLICATION •Manufacturing •Patient Therapy •Side Effect Monitoring •Formulation Enhancement •Phase 1- Human Safety & pK •Phase 11- Human Efficacy •Phase 111- Pivotal Large Scale Efficacy Studies This Means That… New candidates are found during the drug discovery phase They enter clinical development, and if approved by EMEA or FDA they become drug products suitable for use in patient therapy The later stages i.e. development & clinical application impose stringent drug-like requirements on the properties of candidates Thus it is necessary to anticipate these requirements during drug discovery & promote exclusively those molecules that have the highest chances of success to the development phase This Unit Focuses On Discovery. This Stage May be Further Sub-Divided: EXPLORATION Understand target & screen for hits LEAD SELECTION Pick diverse leads • Biological Target • In vitro enzyme & (ID Validation & Characterisation) •Chemical Libraries •HTP Screening •Hit Selection Receptor Assay • Initial in vitro SAR • Property Screens • Initial Synthetic Enhancement LEAD OPTIMISATION Best SAR; SPR Least Side Effects • In vivo SAR • Selectivity Assays • X Ray & NMR Binding Studies • Computational Modelling • Custom Property Studies for SPR • In vitro pK & Metabolism • Analog Synthesis DEVELOPMENT SELECTION Meet Advancement Criteria • Synthetic Batch Scale-up • In vitro & In vivo toxicity •Formulation •In-Depth Property Characterisation •Clinical Candidate Advancement Pick Diverse Leads..... If the consensus is that leads are ligands that typically exhibit suboptimal target binding affinity.. In this scenario acceptable leads must possess very specific characteristics if they are to be considered for further development: Lead Molecules Must: Have relatively simple chemical features Subscribe to a well established SAR series Enjoy a favourable patent situation Possess good SPR characteristics i.e. good ADME/tox properties Identification of Molecules Involves: Such Casting a broad net that pharmacophoric structural space Narrowing possibilities such selected explores Lead diverse that only a few are Carrying out lead optimisation. This is the structural modification of lead molecules to explore SAR. Candidates selected for development subjected to indepth studies which qualify/disqualify them for further development THIS REPRESENTS A CHANGE IN STRATEGY FROM OLDER DRUG DESIGN METHODS…. Early drug design protocols focused on the isolation of active compounds Issues such as pK, toxicity & solubility were addressed much later in the development phase A landmark paper published in the British Journal of Clinical Pharmacology by Prentis et al. in 1988 showed that drugs failed in the development phase for precisely this reason… Prentis et al. SHOWED THAT… ∼39% of drugs failed during the development phase due to poor biopharmaceutical properties This represented a major economic loss for pharmaceutical companies Thus biopharmaceutical properties are more correctly addressed during the discovery phase This would ensure that outright failures are eliminated early in the discovery phase Prentis, R.A., Lis, Y., & Walker, S.R. (1988). Pharmaceutical Innovation by the Seven UK-owned Pharmaceutical Companies (1964-1985). British Journal of Clinical Pharmacology, 25, 387-396 Medicinal Chemistry Space Related to Drug Discovery Chemical Space Druglike Leadlike THE REALITY IS… That drugs with marginal properties (poor solubility & stability) still make it to the development phase This still contributes to the general inefficiency of the drug discovery process MORE ALARMINGLY the burden of mediocre drug properties is sometimes shifted to patients: Poor drug absorption requires dose increases in order to attain therapeutic levels Dosage regimens may require increased frequency of administration or worse change in route from oral to parenteral (considered unacceptable for dosing among wide patient populations) Short t1/2 due to metabolic instability may also require increased frequency of administration at the expense of patient compliance) IN SUMMARY….. It is better to improve the drug-like properties of a molecule during the discovery phase This is best accomplished by changing the chemical structure Modifications are generally made at sites shown by SAR studies as non-critical to therapeutic binding When this is not possible, drug molecules though clinically promising, should be discarded STRESS ON CHANGE IN FOCUS IN CONTEMPORARY DRUG DESIGN STRATEGY PAST CONTEMPORARY Focus on binding affinity to active site Followed by exploration of SAR and.. Optimisation through analog synthesis around core structural scaffold PRESERVATION OF DRUG-LIKE PROPERTIES OF PARAMOUNT IMPORTANCE REALISATION THAT DRUG DESIGN STUDIES BASED ON AFFINITY & POTENCY ALONE INHERENTLY FLAWED……….. Candidates may be too polar to penetrate BBB and reach CNS targets Candidates may be unstable & rapidly cleared through first pass metabolism Candidates may be too soluble to be absorbed from the intestine COMPARING GRAPHICALLY……………… Properties GOOD DRUG GOOD LIGAND Activity THUS… If the focus of a drug design study is solely activity then this may yield compounds that are effective ligands for the target site but which have inadequate properties that would make them successful drugs FOR EXAMPLE… Increasing lipophilicity may increase target protein binding at the expense of aqueous solubility & metabolic stability THUS… A holistic approach that balances attention between activity and physicochemical properties is more likely to yield candidates that can become good drugs AND… The most active or compound may not make drug product because of limitations such as poor pK profile selective the best property or safety AND CONVERSELY….. A less potent compound with better properties may produce better in vivo therapeutic response and prove to be a better product for patients MORE SIMPLY: There are a number of hurdles which a drug molecule must overcome Juggling or simultaneously monitoring and balancing an ensemble of crucial elements is vital to ultimate success Neglecting even one element may cause the entire ensemble to crash ADVANTAGES OF PROPERTY OPTIMISATION INCLUDE... Better planning, execution & interpreting of discovery experiments Reduced discovery time lag from not having to fix property-based problems at a later time Faster & more economical pharmaceutical development Candidates with lower risk and higher future value Longer patent life Higher patient acceptance and compliance BARRIERS TO DRUG EXPOSURE IN LIVING SYSTEMS Physiological barriers reduce the amount of dosed compound that reaches the target Physiological barriers include membranes, pH, metabolic enzymes, & transporters Good properties facilitate good absorption, distribution, low metabolism, reasonable elimination & low toxicity IN VIVO.. Drugs encounter many barriers from the point of administration up to the time it reaches its therapeutic target Thus, besides the inherent affinity that a drug has for a therapeutic target, its ability to overcome these barriers determines the in vivo efficacy of the drug PHYSIOLOGICAL BARRIERS: When a drug molecule encounters a barrier, the amount of drug reaching the other side is diminished: BARRIER DRUG TARGET THUS…. The penetration of drugs to the therapeutic target is slowed and attenuated by the barrier AND… How molecules behave at each barrier determines the rate at which molecules progress to the target site Optimisation of the behaviour of the molecule at the barrier sites results in the molecule arriving at the target at higher concentrations which may lead to the desirable sustainable efficacy A DIVERSE ensemble of physicochemical & biochemical processes is encountered by drug molecules.. Cell Membranes Metabolic Enzymes Solution pH Efflux Transporters Binding Proteins Efficacy…. Is a function of the molecule’s inherent affinity for the target site Is a function of the exposureconcentration & duration of the molecule to the target site Thus the process of drug discovery is representative of a search for molecules possessing structural features that produce: Strong target binding using Structure Based Drug Design (SBDD) and Structure Activity Relationship (SAR) High performance at in vivo barriers using property based design and the Structure Property Relationship (SPR)* * Van de Waterbeemd, H., Smith, D.A., Beaumont, K. & Walker, D.K., (2001) Property Based Design: Optimisation of Drug Absorption & Pharmacokinetics. Journal of Medicinal Chemistry, 44, 13131333 SPR STUDIES: PROPERTY IMPLICATION A molecule’s physicochemical properties The behaviour of a drug molecule in solution & at membrane barriers A molecule’s binding to and reaction with, specific enzymes How the molecule behaves at metabolic barriers A molecule’s binding to various transporters & plasma proteins The absorption, distribution & excretion of the drug A molecule’s reactivity & binding Toxicity DRUG DOSING A common goal of pharmaceutical researchers is the development of a drug dosage form that: Is a low dose tablet Has an oral once daily dosage regimen IDEALLY SUCH A DRUG PRODUCT: Has reasonable manufacturing & storage costs Attracts high patient compliance COMPOUNDS with limited performance at one or more in vivo barriers: May have poor pK performance Require adjustment from the ideal strategy previously outlined May require more frequent dosing (if t1/2 is short) May require higher dosage (if bioavailability is low) May require administration through an alternative route (e.g. i.v.) (if absorption is low) May require a different vehicle or formulation (if solubility is low) A TRADEOFF CONSEQUENTLY EXISTS… Between structural features that enhance therapeutic drug binding AND…. Structural features that enhance delivery through optimal performance at in vivo barriers CONSEQUENTLY… If a drug discovery program focuses exclusively on activity optimisation Poor drug properties may arise POOR DRUG PROPERTIES MAY INCLUDE… Low absorption- low solubility or permeability High clearance- owing to metabolism High clearance by hydrolysis- in GIT or blood Efflux- opposes uptake in many membranes which enhances extraction in liver & kidney High protein binding- limits free drug at target Poor penetration of a blood-organ barrier at the target organ High volume of distribution due to lipophilicity ALL MAY BE CHEMISTS MODIFICATION IMPROVED THROUGH BY MEDICINAL STRUCTURAL RULES GOVERNING RAPID PROPERTY PROFILING FROM STRUCTURE The application of rules provide a rapid method through which the drug-like properties of a compound may be evaluated These rules are a set of guidelines for the structural properties of compounds that have a higher probability of being well absorbed after drug dosing These rules are widely embedded in drug modeling software packages IMPORTANT CAVEATS These guidelines are not absolute They do not intend to form strict cutoff values to categorise between drug-like and non-drug-like molecules Nonetheless, they have been found to be quite effective and efficient to apply LIPINSKI RULES Medicinal chemists and pharmaceutical scientists have used structural properties in various ways for many years Rules became more prominent and defined in the field with the report by Lipinski et al.* of the Rule of 5, or as they are better known the Lipinski Rules These rules are a set of property values that were derived from classifying the key physicochemical properties of drug-like compounds These rules were used at Pfizer for a few years prior to their publication Since then they have become very widely used *Lipinski, C.A., Lombardo, F., Donimy, B.W., & Feeny, P.J. (1997). Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Advanced Drug Delivery Reviews, 23, 3-25 THE IMPACT OF THESE RULES IN THE FIELD HAS BEEN HIGH… Fast, easy & no cost to implement 5 mnemonic makes them easy to remember Intuitively evident to medicinal chemists Widely used standard benchmark Based on solid research, documentation and rationale Work effectively LIPINSKI’S ARTICLE STATES.. Poor absorption or permeation is more likely when: 1. 2. 3. 4. 5. There are more than 5 hydrogen bond donors (expressed as the sum of all OH and NH groups) MWt greater than 500 logP greater than 5 There are more than 10 hydrogen bond acceptors (expressed as the sum of all Ns and Os) Substrates for biological transporters are exceptions to this rule EXPERIENCE SHOWS THAT… Violation of one rule may not necessarily result in poor absorption However, likelihood of poor absorption increases with the number of rules broken and the extent to which they are exceeded HOW WERE THE LIPINSKI RULES DERIVED? Examination of the structural properties of compounds which had survived Phase 1 clinical trials and had moved on to Phase 11 studies Phase 1 studies involve human dosing to determine toxicity and pharmacokinetics The fact that they had moved on to Phase 11 studies implied that these compounds were sufficiently well absorbed in humans for pharmaceutical companies to continue investing in their development A set of 2200 compounds was examined, and the clear trends that were observed became the basis for these rules THE RULES WERE SET AT THE 90th PERCENTILE OF THE COMPOUND SET This means that 90% of the compounds that had sufficient absorption after oral dosing had molecular property values within the Lipinski Rules Compounds that approach or exceed these values have a higher risk of poor absorption after oral dosing THE RULES ARE BASED ON A STRONG PHYSICOCHEMICAL RATIONALE….1 Hydrogen bonds increase solubility in water and must be broken in order for a compound to permeate the lipid bilayer membrane Thus increasing the number of hydrogen bonds reduces partitioning from the aqueous phase into the lipid bilayer membrane for permeation by passive diffusion THE RULES ARE BASED ON A STRONG PHYSICOCHEMICAL RATIONALE….2 Molecular Weight is related to the size of the molecule As molecular size increases, a larger cavity must be formed in water to solubilise the compound thus decreasing solubility Increasing size also impedes passive diffusion through the tightly packed aliphatic side chains of the lipid bilayer membrane THE RULES ARE BASED ON A STRONG PHYSICOCHEMICAL RATIONALE….3 Increasing logP decreases aqueous solubility which reduces absorption Membrane transporters can either enhance or reduce compound absorption by either active uptake transport or efflux respectively This means that transporters can have a strong impact on increasing or decreasing absorption LIPINSKI et al. …. In their paper discuss the important implications of these rules in the light of current drug discovery strategies: The discovery lead optimisation stage often increases target binding by adding hydrogen bonds and increasing lipophilicity This means that activity optimisation may reduce the drug-like properties of a compound series THE LIPINSKI RULES are widely used as a filter and measurement of the drug-likeness of a series of molecules. They are used to such an extent that they almost ‘copyright’ the field of drug- likeness compound scoring. However, other experiments have also been carried out in this area….. VEBER RULES The results of an experiment performed by Veber et al.* examining the oral bioavailability of potential drug candidates in the rat let to the conclusion that other parameters existed for the description of drug likeness than the Lipinski rules. The main parameter taken into account during this experiment was the number of rotatable bonds – as an indication of molecular flexibility. *Veber, D.F., Johnson, S.R., Cheng, H., Smith, B.R., Ward, K.W., & Kopple, K.D. (2002). Molecular Properties that Influence the Oral Bioavailability of Drug Candidates. Journal of Medicinal Chemistry, 45, 2615-2623 VEBER RULES Veber’s experiments indicated that the main factor influencing the possibility of uptake by the lumen is not molecular weight but, in fact the number of rotatable bonds. This could be explained by the entropic cost of presenting an acceptable drug surface area to hydrophobic surface of the membrane in the sense that a compact molecule is easier to absorb than extended one. In addition to the number of rotatable bonds Veber et al. found that the polar surface area can be used as a good indication of permeation. Crossing the lumen requires for a molecule that it is rather non-polar, and therefore having a large polar area as part of the surface makes the interaction and uptake over a lipid bilayer difficult. VEBER RULES They therefore suggest the following filter for drug-likeness: Rotatable bonds < 12 Polar surface area < 140 Also, Veber et al. (2002) therefore raise the issue of molecular weight being a proper descriptor for absorption measurement as molecular weight might just be positively correlated with more precise properties like the rotatable bonds count, polar surface area and hydrogen bonds count. The Veber et al. experiments referred to above underline the difficulties met with when trying to make generalizing rules for complex systems. CORRESPONDENCE BETWEEN MOLECULAR WEIGHT, THE NUMBER OF ROTATABLE BONDS AND THE DEGREE OF BIOAVAILABILITY IN THE RAT APPLICATION OF RULES FOR COMPOUND ASSESSMENT TO ANTICIPATE THE DRUG LIKE PROPERTIES OF COMPOUNDS WHEN PLANNING SYNTHESIS TO ESTIMATE THE DRUG LIKE PROPERTIES OF HITS FROM HIGH THROUGHPUT SCREENING TO EVALUATE THE DRUG LIKE PROPERTIES OF COMPOUNDS BEING CONSIDERED AS CANDIDATES FOR PURCHASE FROM A VENDOR This Philosophy Recalls the Adage: The house built on a foundation (LEAD STRUCTURE) of sand will fall, but the house built on rock will prosper In the Drug Discovery Process: If the lead molecule (foundation) is strong, the project team can build a strong drug-like clinical candidate If the lead molecule (foundation) is weak, the team’s effort may never advance a drug-like compound to development Hits That Serve as Starting Points for Leads Come From.. High Throughput Screening Virtual Screening Natural Ligands Natural Products Scientific Literature Hit-to-Lead Phase- Emerging Concepts Lead likeness Template Conservation Triage Fragment-based Screening Lead Likeness.......................1 Was initially based on Rule of 5 (invaluable as property guidelines in lead selection) Implication was that the leads (foundation) would be free of major liabilities that would later impede viability as clinical candidates Lead Likeness.......................2 With acquired experience it was recognised that during lead optimisation often substructures were added to lead templates to enhance target affinity and selectivity: Non-polar groups added to enhance binding to lipophilic pockets Polar groups added to increase hydrogen bonding to binding site Lead Likeness.......................3 This process often resulted in Rule of 5 violators with deleterious properties Current philosophy advocates that screening libraries select leads with: 1. Molecular weight between 100 & 350 2. ClogP between 1 & 3 This increases the odds that the optimisation process results in molecules with acceptable drug-like properties Lead Likeness.......................4 Another caveat was that these curtailed optimised lead molecules would be more likely to bind to target proteins owing to the fact that it is easier for them to adopt bioactive conformations than their larger counterparts which are commonly included in screening libraries Lead Likeness.......................5 Criteria for Inclusion to Lead-Like Screening Libraries MW ≤460 Log P -4≤Log P≤4.2 Log Sw ≤-5 Rotatable Bonds ≤10 Rings ≤4 Hbond Donors ≤5 Hbond Acceptors ≤9 Pharmacokinetic Criteria for Inclusion to Lead-Like Screening Libraries Bioavailability (%F) ≥30% Clearance (Cl) ≤30ml/min/kg in rat Log D 0≤Log D7.4≤3 Cytochrome P450 Binding Low Plasma Protein Binding ≤99.5% Acute & Chronic Toxicity None (in therapeutic window) Template Conservation..........1 Often a large portion of lead structure is conserved throughout lead optimisation Lead optimisation is synonymous with structural additions Properties associated with the core continue to be a primary component of the properties of analogs and of the eventual clinical candidate Template Conservation..........2 O N Screening Lead S O N O Liranaftate S O H Natural Product Lead H O O Exemestane H O H Many drugs retain large portions of lead core structures Implication is that it is sensible to lock in favourable properties at the lead selection stage Template Conservation..........3 Going back to the analogy with houses and foundations, attempts to endow non-drug-like leads with drug-like properties, would be similar to attempting to reconstruct the foundations of a house that has already been built. Complexity, time and financial intensiveness would make such an exercise unfeasible Triage..................................1 Early screening in a drug design project normally yields many hits for consideration Inclusion of property characteristics with activity, selectivity & novelty represents a sound strategy that ensures strong leads i.e. Those with the greatest chance of success at the expense of those with high failure risk which are consequently downgraded Triage..................................2 Sets goals for each key criteria of the lead Disciplined process Guides initial synthetic modifications for improvement & selection of leads for optimisation LEAD ANALOG DESIRED PROFILE MW 330 445 <450 clogP 1.9 5.19 <4.0 IC50 (µM) 4.2 >20 <1.0µM Binding to Target X-ray Yes MIC B. subtilis >200µM 50µM <200µM S. aureus MRSA >200µM 25µM <200µM S. aureus ATTC >200µM 200µM <200µM S. pneumo + >200µM 25µM <200µM Selectivity: C. albicans (MIC µg/ml) >200 >200 >10 fold Aqueous Solubility (µg/ml @ pH 7.4 >100 26.5 >60 0 0.15 >1 CYP3A4 (%inhibition @ 3µM) 11 7 <15 CYP2D6 (%inhibition @ 3µM) 0 1 <15 CYP2C9 (%inhibition @ 3µM) NT 23 <15 Microsome stability (% remaining @30 mins) NT NT >80 Definable series Yes Yes Yes Definable SAR Yes Yes Yes Permeability (10-6 m/s @ pH 7.4) Example of goals used by Wyeth Research exploratory medicinal chemists for hit selection, initial structural modification & lead selection in an acyl carrier protein synthase (AcpS) inhibitor project Fragment-Based Screening.....1 Based on the theory that screening with larger structures that fit (shape, electrostatic interactions, & hydrophobic contacts) the binding site of the target does not constitute Good Drug Discovery Practice The use of smaller, less complex compounds or fragments is more likely to bind to a portion of the binding site Fragment-Based Screening.....2 From a fragment core, functionality can be added to enhance binding. By selecting fragments that bind to different portions of the site and joining them together with a tether the likeliehood of finding a final lead that binds appreciably to the site increases. Although fragments bind with low affinity, tethered fragments forming a larger molecule normally bind with a greater affinity. Fragment-Based Screening.....3 When fragments bind they generally have a lower affinity (IC50 50µM-1µM) but have a high efficiency of binding for their size. Fragment binding is difficult to detect using biological assays, but X-ray crystallography and NMR may be used to resolve such weakly binding fragments This is important in ligand binding pocket orientation determination Disadvantage: Expense Excellent reviews published Lesuisse, D., Lange, G., Deprez, P., Bernard, D., Schoot, B., Delettre, G., et al. (2002). SAR and X-Ray. A New Approach Combining Fragment Based Screening and Rational Drug Design: Application to the Discovery of Nanomolar Inhibitors of Src SH2. Journal of Medicinal Chemistry, 45, 2379-2387. Fragment-Based Screening.....4 Complements the goal of selecting leads with good properties. When screening large molecules (common in conventional screening libraries), often, considerable portions of the structure are uninvolved in binding interactions All that is achieved is an increase in MW, hbonds and lipophilicity that detract from the lead-like properties. Fragment-Based Screening.....5 Fragment use can minimise superfluous structural moieties that would detract from optimum absorption profiles. Fragment libraries are in fact now being constructed exclusively from these small molecules, which already have good lead-like properties. Fragment-Based Screening.....6 This has lead to the establishment of guidelines for properties which molecules must possess in order to be included in fragment libraries Property Value MW ≤300 ClogP ≤3 Rotatable Bonds ≤3 Hbond Donors ≤3 Hbond Acceptors ≤3 PSA ≤60Å2 Lipophilicity Tendency of compound to partition into a nonpolar liquid matrix versus an aqueous one Estimated using logP from octanol/water partitioning Lipophilicity is major determinant of many ADME/Tox properties & of pharmacological activity It can be quickly measured or calculated thanks to work of Hansch & Leo (advantage) Its inclusion into the Rule of 5 indicates its effectiveness in initial compound assessment Hansch, C., Leo, A., & Hoekman, D., (1995). Exploring QSAR. Fundamentals and Applications in Chemistry & Biology, Volume 1. Hydrophobic, Electronic & Steric Constants, Volume 2. New York: Oxford University Press Traditionally Lipophilicity Assessed: By partitioning compound between nonpolar (octanol) and polar (water) phases Partitioning values measured are LogP and Log D LogP Log of partition coefficient of the compound between an organic phase and an aqueous phase at a pH when all of the compound molecules are neutral: Log P=log ([Compoundorganic]/[Compoundaqueous]) Depends on partitioning of molecules between 2 matrices Log D Log of distribution coefficient of the compound between an organic phase and an aqueous phase at a specific pH(x). A portion of the compound molecules may be in the ionic form and a portion may be in the neutral form: Log DpHx=log ([Compoundorganic]/[Compoundaqueous]) LogD Depends on partitioning of the neutral portion of the molecule population plus the partitioning of the ionised portion of the molecule population. Ions have greater affinity for the polar aqueous phase than for the nonpolar organic phase. Fraction of ionised molecules is dependent on pH of the aqueous solution, the pKa of the compound, and on whether it is an acid or a base. LogD For acids, the neutral/anion ratio of molecules in solution decreases with increasing pH. Thus logD decreases with increasing pH. For bases, the neutral/anion ratio of molecules in solution in.creases with increasing pH. Thus logD increases with increasing pH. LogP Abraham et al. showed that logP is affected by several fundamental structural properties of the compound: 1. Molecular Volume 2. Dipolarity 3. Hydrogen Bond Acidity 4. Hydrogen Bond Basicity Abraham, M.H., Chadha, H.S., Leitao, R.A.E., Mitchell, R.C., Lambert, W.J., Kaliszan, R., et al. (1997) Determination of Solute Lipophilicity, as logP(octanol) and log P(alkane) Using Poly(styrene-divinylbenzene) and Immobilised Artificial Membrane Stationary Phases in Reversed Phase High Performance Liquid Chromatography. Journal of Chromatography A, 766, 35-47 Log P Molecular Volume: related to molecular weight & affects the size of the cavity that must be formed in the solvent to solubilise the molecule Dipolarity: affects the polar alignment of the molecule with the solvent Hbond Acidity: related to hbond donation Hbond basicity: related to hbond acceptance Lipophilicity Changes with the condition of the phases: 1. 2. 3. 4. 5. Partitioning solvents pH Ionic strength Buffer Co-solutes/Co-solvents Lipophilicity Partitioning between octanol and water different to that between cyclohexane and water due to differences in the molecular properties of the phases pH affects degree of ionisation Increasing ionic strength results in increasing polarity of the aqueous phase The buffer also affects polarity, molecular interactions and formation of in situ salts as counterions with drug molecules Co-solvents such as DMSO can interact with solutes & change their partitioning behaviour Lipophilicity Generally optimal GI absorption by passive diffusion after oral dosing is to have a moderate LogPrange 0-3 In this range, a good balance of permeability and solubility exists Compounds with a lower LogP are more polar and have poorer lipid bilayer permeability Compounds with a higher LogP are more non-polar and have poor aqueous solubility LogD7.4 Common Impact on Drug-like Properties Common Impact in vivo <1 High Solubility Low Permeability by passive diffusion Paracellular permeability possible if MW <200 Vd low Oral absorption and BBB penetration unfavourable Renal Clearance may be high Solubility moderate Permeability moderate Balanced Vd Solubility Low Permeability high Metabolism moderate to high Oral bioavailability moderate to low Oral absorption variable Solubility low High Vd especially amines Oral absorption unfavourable & variable 1-3 3-5 >5 Permeability high Metabolism high Oral absorption and BBB penetration unfavourable LogD7.4 <1: good solubility but low absorption or brain penetration owing to low passive diffusion permeability . 1 < LogD7.4 <3: This is an ideal range. These compounds generally have good intestinal absorption owing to a good balance of solubility and passive diffusion permeability. Metabolism is minimised owing to lower binding to metabolic enzymes . 3 < LogD7.4 <5: These compounds have good permeability but lower absorption due to low solubility. Metabolism is increased in this range, owing to increased binding to metabolic enzymes. LogD7.4 <5: Compounds in this range tend to have low absorption and bioavailability, owing to low solubility. Metabolic clearance is high because of high affinity for metabolic enzymes. Vd and half life are high because compounds partition into, and stay in tissues ∆logP Is used to predict permeation of BBB It is the logP from partitioning between octanol and water, minus the logP from partitioning between cyclohexane and water. The difference is attributed to the contribution of hbonding to log Pow (octanol/water) compared to log Pcw (cyclohexane/water) As ∆logP increases, BBB permeability generally decreases. Its correlation to BBB permeation has been interpreted in terms of hbonding on BBB permeability pKa The ionisability of a compound is indicated by its pKa Ionisability is a major determinant of solubility and permeability When pH=pKa, the concentration of ionised and neutral molecules in solution are equal Basicity of bases increases as pKa increases; acidity of acids increases as pKa decreases pKa The great majority of drugs contain ionisable groups Most are basic Some are acidic Only 5% are not ionisable Medicinal chemists can modify acidic or basic substructures in order to obtain the desired pKa which affects solubility & permeability pKa Fundamentals pKa: negative log of the ionisation constant Ka It is common to use pKa for both acids and bases For Acids: HA=H++ApKa=-log([H+][A-]/[HA]) For Bases: HB=H++BpKa=-log([H+][B]/[HB+]) Therefore: For acids, as pH decreases there is a greater concentration of neutral acid molecules (HA) and a lower concentration of anionic acid molecules (A-) in solution Acids with a lower pKa are strongerwith a greater tendency to form A- And Therefore: For bases, as pH decreases there is a lower concentration of neutral base molecules (B) and a higher concentration of cationic base molecules (HB+) in solution Bases with a lower pKa are weakerwith a lower tendency to form HB+ The Henderson-Hasselbach Equation: Is a useful relationship for discovery: For acids: pH=pKa+log([A-]/[HA]) or [HA]/[A-]=10(pKa-pH) For bases: pH=pKa+log([B-]/[HB+]) or [BH+]/[B]=10(pKa-pH) These relationships provide a means of calculating the concentration of ionic and neutral species at any pH if pKa is known. Moreover: Moreover, it is useful to note that when pH=pKa, then there is an equal concentration of ionic and neutral species in solution. Pka Effects... Ionised molecules are more soluble in aqueous media than neutral molecules because they are more polar Solubility is determined both by the intrinsic solubility of the neutral molecule and that of the ionised species which is much greater Conversely: Ionised molecules are less permeable than neutral molecules The neutral molecules are more lipophilic than their ionised counterparts and are considered to be the dominant form that permeates by passive diffusion Pka Effects... Since pKa determines the degree of ionisation, it has a major effect on solubility and permeability. These in turn determine intestinal absorption after oral dosing. Thus, highly permeable compounds often have low solubility and vice versa. Thus there is a tradeoff between solubility and permeability because of the opposite effects of ionisation on these properties For Example: An acidic compound with a pKa of 5 exhibits decreasing permeability as the pH of the solution increases. Conversely, the solubility increases at increasing solution pH. pKa also affects the activity of a structural series. This is due to changes in interactions at the active site of the target protein pKa Case Studies Bases pKa Guanidine 13.6 Acetamide 12.4 Pyrrolidine 11.3 Piperidine 11.1 Acids pKa Methylamine 10.6 CF3COOH 0.23 Piperazine 9.8;5.3 CCl3COOH 0.9 Trimethylamine 9.8 CCl2HCOOH 1.3 Glycine 9.8 CClH2COOH 2.9 Morpholine 8.4 HCOOH 3.8 Imidazole 6.8 C6H5COOH 4.2 Pyridine 5.2 Succinic Acid 4.2;5.6 Quinoline 4.9 H3COOH 4.8 Aniline 4.9 Thiophenol 6.5 Triazole 2.5 p-Nitrophenol 7.2 Purine 2.4 m-Nitrophenol 9.3 Pyrimidine 1.2 C6H5OH 10.0 Diphenylamine 0.8 Drugs & pKa Acids pKa Penicillin V 2.7 Salicylic Acid 3.0;13.8 Acetylsalicylic Acid 3.5 Diclofenac 4.1 Sulfathiazole 7.1 Phenobarbital 7.4;11.8 Phenytoin 8.3 Acetaminophen 9.9 Caffeine 14 Bases pKa Caffeine 0.6 Quinidine 4.1;8.0 Tolbutamide 5.3 Cocaine 8.4 Ephedrine 9.4 Imipramine 9.5 Atropine 9.7 In Summary: When synthetic modifications are planned for the purpose of improving the water solubilty or permeability of a structural series, a wide selection of substructures can be used. It is important to remember that structural modifications that increase solubility will also decrease permeability In Summary: By modifying the substructures of a molecule to introduce groups with differing pKa values, medicinal chemists can modify the solubility and permeability of the compound