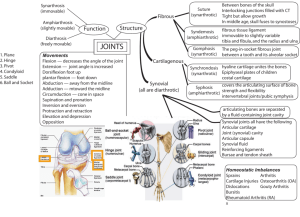
Expression of Alpha-Smooth Muscle Actin and... Collagen-Glycosaminoglycan Scaffolds by Cells
advertisement

Expression of Alpha-Smooth Muscle Actin and Contraction of Collagen-Glycosaminoglycan Scaffolds by Cells Derived from Canine Synovium By Scott M. Vickers B.S., Mechanical Engineering University of Kentucky, 2001 Submitted to the Department of Mechanical Engineering in Partial Fulfillment of the Requirements for the Degree of Master of Science in Mechanical Engineering at the Massachusetts Institute of Technology MASSACHUSETTS INSTITUTE OFTECHNOLOGY February 2003 JUL 0 8 2003 LIBRARIES @ 2003 Massachusetts Institute of Technology All rights reserved Signature of Author: Department of Mechanical Engineering January 14, 2003 Certified by: // SV * /Myron Spector Senior Lecturer, Department Mech/ ical Engineering Professor of Orthopaedic Surgery (Biomateri ), Harvard Medical School Thesis Supervisor Accepted by: ''.... Ain Al Sonin Chairman, Department Committee on Graduate Students 1000:1 40C Expression of Alpha-Smooth Muscle Actin and Contraction of Collagen-Glycosaminoglycan Scaffolds by Cells Derived from Canine Synovium By Scott M. Vickers Submitted to the Department of Mechanical Engineering on January 14, 2003 in Partial Fulfillment of the Requirements for the Degree of Master of Science in Mechanical Engineering ABSTRACT Recent studies have demonstrated that several types of musculoskeletal connective tissue cells - including chondrocytes, fibrochondrocytes, ligament fibroblasts, osteoblasts, and mesenchymal stem cells - can express the gene for the contractile actin isoform, alpha-smooth muscle actin (SMA), and can contract analogs of extracellular matrix. While the physiological role of the SMA-enabled contraction of these cells remains to be elucidated, such contractility may have detrimental effects when the cells are seeded in scaffolds employed for tissue engineering. These prior findings prompted investigation of SMA expression in synovial cells. These cells have attracted recent interest as donor cells for tissue engineering of articular cartilage because they have been implicated in certain cartilage repair processes in vivo and the chondrogenic potential of the cells has recently been demonstrated in vitro. The objective of this study was to evaluate SMA expression by adult canine synovial cells and their related contraction of collagen-glycosaminoglycan (GAG) analogs of extracellular matrix, as well as cellular proliferation and chondrogenic potential within the scaffolds. Cells from synovial membranes of 6 adult dogs were isolated by outgrowth from the tissue and expanded through seven passages in monolayer culture, with samples from each passage allocated for Western blot analysis of SMA. Cells from passage 4 were seeded into porous type I collagen-GAG matrices and cultured for 4 weeks. The diameters of the cell-seeded scaffolds and non-seeded controls were measured every other day. Synovium-derived cells from the fourth passage were formed into micro-pellets by centrifugation and others seeded into the collagen-GAG matrices, and were incubated in chondrogenic medium with and without fetal bovine serum. After 3 weeks the specimens were prepared for type II collagen immunohistochemistry. Immunohistochemistry revealed the presence of SMA in some cells in the intimal layer of synovium from 4 of the 5 animals analyzed. Western blot analysis demonstrated a regular increase in the amount of SMA in the synovial cells with passage number. The synovial cellmediated contraction of the collagen-GAG scaffolds reached a value of 43% of the original diameter after 4 weeks. This cell-mediated contraction associated with SMA expression was comparable to that found with other musculoskeletal cell types. Incubation of cultures of synovial cells with chondrogenic medium revealed trace amounts of type I collagen production by immunohistochemistry, suggesting that they may be capable of differentiating into chondrocytes. The findings of this study indicate that control of SMA-enabled contraction may be important when employing synovial cells for cartilage repair procedures, and warrant further investigation into the physiological role of SMA expression in synovial cells. Thesis Supervisor: Myron Spector Title: Senior Lecturer, Department of Mechanical Engineering Professor of Orthopaedic Surgery (Biomaterials), Harvard Medical School 3 ACKNOWLEDGEMENTS The work presented here could not have been accomplished without the help and guidance of a great number of people, to whom I am deeply indebted. First, I must thank my supervisor, Prof. Myron Spector, for the opportunity to work on a project from which I have learned so much. Your guidance, support, encouragement, and concern for my development as a researcher and scholar are greatly appreciated. Your enthusiasm is contagious, and has greatly stimulated my interest in the field of tissue engineering. Secondly, I would like to thank Prof. Yannas and Prof. Gibson for their advice and the use of their laboratory space and equipment. You have both taught me a great deal. In addition, I must thank all of the individuals who have been a part of the Orthopaedics Research Laboratory at Brigham and Women's Hospital and the Fibers and Polymers Laboratory at MIT for your assistance in so many areas. " Brendan, thank you for coming in at odd hours to teach me to produce matrices, for help with classes, and for your encouragement in general. * Dr. Hsu and Dr. Xiang, thank you for teaching me to harvest synovium, and for the many trips you made with me to NEMC. " Robyn, thanks for teaching me to do Western blots, DNA analysis, and general cell culture procedures. " Liqun, thank you for helping me improve my Western blot protocols, and for running so many of the assays when I was pressed for time. " Nikki, Ramille, Dan, Leonide, Dawn, Jamil, Tim, Changming, and Ricardo, thanks for your everyday companionship, stimulating discussions, and for making the lab such an enjoyable workplace. The financial support for this study provided by the Cambridge-MIT Institute is gratefully acknowledged. To all of my family and friends who have supported me throughout this entire process, I cannot thank you enough. Luke, thanks for keeping me on my toes, and making my life so much fun. And finally, Jenny, the acknowledgment you deserve cannot be adequately put into words. Thank you for your companionship in all of life, for supporting me in my academic endeavors, for sharing my times of excitement and sympathizing with my frustrations. Thanks for taking care of the details, and for always being there. 4 TABLE OF CONTENTS ACKNOW LEDGEM ENTS ...................................................................................................... 4 TABLE OF CONTENTS ........................................................................................................ 5 LIST OF FIGURES ....................................................................................................................... 7 1. INTRODUCTION AND BACKGROUND......................................................................... 9 1.1 CLINICAL PROBLEM: CARTILAGE DAMAGE AND DEGRADATION .................................... 1.2 CURRENT CLINICAL TREATMENTS.................................................................................. 9 10 1.2.1 Microfracture Technique ....................................................................................... 1.2.2 Tissue and Cell Transplantation........................................................................... 10 11 1.3 PROSPECTS FOR TISSUE ENGINEERING............................................................................ 12 1.4 REVIEW OF SYNOVIUM, COLLAGEN-GAG SCAFFOLDS, AND SMA-ENABLED CO N TR A CTION ................................................................................................................. 13 1.4.1 Synovium Structure and Function......................................................................... 1.4.2 Collagen-GAG matrices......................................................................................... 1.4.3 SMA-Enabled Contraction..................................................................................... 13 14 14 2 SPECIFIC AIM S AND W ORKING HYPOTHESES..................................................... 15 3 MATERIALS AND METHODS ....................................................................................... 17 3.1 SYNOVIAL CELL ISOLATION AND CULTURE................................................................... 3.2 THREE-DIMENSIONAL CELL CULTURE USING COLLAGEN-GAG SCAFFOLDS................ 17 18 3.2.1 Matrixfabrication.................................................................................................. 18 3 .2 .2 Cell-Seeding ............................................................................................................... 18 3.2.3 Matrix ContractionMeasurements ......................................................................... 18 3.3 3.4 3.5 3.6 3.7 CHONDROGENIC DIFFERENTIATION CULTURE ................................................................. W ESTERN BLOT ANALYSIS FOR SMA ............................................................................ D N A A N A LY SIS .................................................................................................................. HISTOLOGY AND IMMUNOHISTOCHEMISTRY .................................................................. STATISTICAL ANALYSIS .................................................................................................. 4 RESULTS ................................................................................................................................ 19 19 20 21 22 23 4.1 SMA EXPRESSION IN VIVO ................................................................................................. 4.2 CELL GROWTH AND IN VITRO EXPRESSION OF SMA ..................................................... 23 25 4.2.1 Cell Growth in Monolayer Culture ....................................................................... 4.2.2 Western Blot Analysis of SMA Content................................................................... 25 25 4.3 CONTRACTION OF COLLAGEN-GAG SCAFFOLDS............................................................... 27 4.3.1 DiameterMeasurements ......................................................................................... 27 4 .3.2 D NA Content .............................................................................................................. 28 4.3.3 Histology of the Cell-SeededMatrices................................................................... 29 4.4 CHONDROGENIC DIFFERENTIATION OF SYNOVIAL CELLS ............................................. 5 DISCUSSION.......................................................................................................................... 5.1 5.2 5.3 5.4 SMA EXPRESSION IN VIVO ............................................................................................. IN VITRO EXPRESSION OF SMA ....................................................................................... CHONDROGENIC DIFFERENTIATION OF SYNOVIAL CELLS ............................................. CONTRACTION OF COLLAGEN-GAG SCAFFOLDS............................................................ 29 35 35 35 36 36 5 6 C ON CLU SIO N S.....................................................................................................................39 7 LIMITATIONS AND FUTURE WORK.......................................................................... 41 REFEREN CES ............................................................................................................................ 43 APPENDIX A: FABRICATION OF COLLAGEN-GAG SCAFFOLDS .............. 49 A . 1 PREPARATION OF COLLAGEN-GA G SLURRY ................................................................. A .2 FREEZE-D RYING ................................................................................................................. A .3 D EHYDROTHERMAL (DH T) CROSS-LINKING .................................................................. 49 50 50 APPENDIX B: CELL ISOLATION AND CULTURE.......................................................... 51 B .1 B .2 B .3 B.4 B .5 TISSUE H ARVEST ................................................................................................................ PASSAGING CELLS .............................................................................................................. CELL COUNTING ................................................................................................................. FREEZING CELLS ................................................................................................................ THAW ING CELLS................................................................................................................. 51 52 53 54 54 B .7 CELL-PELLET CULTURES................................................................................................ 55 APPENDIX C: BIOCHEMICAL ASSAYS............................................................................ 57 C.1 W ESTERN BLOT FOR DETECTION OF SM A ....................................................................... 57 C.1.1 Protein Extraction.................................................................................................. C.1.2 Protein Assay ............................................................................................................. C.1.4 Blot Transfer .............................................................................................................. C. 1.6 D ensitometric Analysis............................................................................................ C.2 DN A ANALYSIS.................................................................................................................. C.2.1 PapainDigestion..................................................................................................... C.2.2 Fluorometric Quantificationof DNA ..................................................................... 57 58 62 64 65 65 65 APPENDIX D: HISTOLOGY AND IMMUNOHISTOCHEMISTRY ............................... 67 D .1 D .2 D.3 D.4 6 PARAFFIN EMBEDDING ....................................................................................................... H EM ATOXYLIN AND EOSIN (H & E) STAINING .................................................................. IMMUNOHISTOCHEMICAL STAINING OF ct-SMOOTH MUSCLE ACTIN............................ IMMUNOHISTOCHEMICAL STAINING OF TYPE II COLLAGEN........................................... 67 68 69 71 LIST OF FIGURES Figure 1: Immunohistochemical staining for cc-smooth muscle actin (SMA) in 2 samples of intact adult canine synovium..................................................... 23 Figure 2: Cell growth rate in monolayer culture.......................................................... 25 Figure 3: SMA content of synovial cells passage number.......................................... 26 Figure 4: Linear regression analysis of SMA content of synovial cells with time in m onolayer culture.......................................................................................... 26 Figure 5: Diameter reduction of synovial cell-seeded and control scaffolds................ 27 Figure 6: Cell-mediated contraction of the collagen-GAG matrices ........................... 28 Figure 7: DNA content of the cell-seeded matrices..................................................... 28 Figure 8: Histology and immunohistochemistry of synovial cell-seeded matrices. ........ 31 Figure 9: Immunohistochemical staining of type II collagen in synovial cell m icropellet and m atrix cultures..................................................................... 33 7 8 1. INTRODUCTION AND BACKGROUND Articular cartilage is a specialized connective tissue that provides a low-friction load-bearing surface in diarthroidal joints such as the knee, hip, and elbow. When damaged, articular cartilage generally does not heal, but rather continues to degenerate, often leading to osteoarthritis. Although several surgical procedures that attempt to induce healing of the damaged cartilage tissue have been reported to relieve symptoms of pain and dysfunction, none have yet achieved the desired regeneration of native cartilage over the long term. Recent studies have indicated the promise of tissue engineering approaches to the problem of cartilage repair and regeneration. Much of the current work has been focused on transplantation of cells from healthy cartilage into the damaged area, with or without a biodegradable scaffold. Recent demonstration of the chondrogenic potential of cells from the synovial membrane, however, has anticipated the use of such cells instead of chondrocytes in cartilage repair procedures, thus eliminating some of the difficulties such as donor site morbidity - encountered when using native chondrocytes. The purpose of this thesis was to lay the groundwork for the development of a tissue-engineered implant employing synovial cells and collagen-glycosaminoglycan (GAG) scaffolds to facilitate articular cartilage regeneration. In designing such implants, it is necessary to understand various aspects of the cell-matrix interaction, such as cellmediated contraction of the construct, as has been reported for a variety of musculoskeletal connective tissue cells. Toward this goal, synovial cell-mediated contraction of the collagen-GAG scaffolds and their related expression of the contractile actin isoform, cc-smooth muscle actin, was examined, as well as cellular proliferation and chondrogenic potential within the scaffolds. 1.1 Clinical Problem: Cartilage Damage and Degradation That spontaneous healing of damaged articular cartilage is rarely encountered has been recognized for over two and a half centuries [25]. Several factors contribute to the limited healing capability of this tissue, including: 1) Articular cartilage is primarily avascular, and thus lacks the systemic supply of cells and soluble regulators that typically initiate the repair process. Additionally, the avascularity precludes the formation of a fibrin clot, which 9 in vascular tissues acts as a temporary scaffold in which cells can migrate and begin synthesizing new tissue. 2) The cell number density in articular cartilage - approximately 10,000 cells/mm3 [50] - is low compared to that of other tissues, thus limiting the number of native cells available to participate in the repair process. 3) The contribution of chondrocytes (cells native to cartilage) to repair of damaged tissue is hindered by their limited migratory, mitotic, and metabolic activity. Instead of healing, lesions in articular cartilage compromise the mechanical properties of the surrounding tissue, predisposing the joint to further degeneration [15, 16]. Such degradation of the joint surfaces can result in the painful and often debilitating condition of osteoarthritis, one of the leading causes of disability affecting nearly 21 million Americans [1]. Severe cases of osteoarthritis often necessitate total joint arthroplasty in order to ameliorate symptoms. While encouraging results of pain relief and improvement of joint function have been reported for joint replacement surgeries in elderly patients, such procedures have a high rate of failure in individuals under the age of 65 [57]. 1.2 Current Clinical Treatments Several surgical procedures focusing on repair of damaged cartilage tissue have been developed in efforts to relieve pain, restore functionality, and avoid or postpone the need for joint replacement in younger individuals (see Hunziker [27] and O'Driscoll [55] for review). Two approaches currently in clinical use are microfracture of the subchondral plate and tissue or cell transplantation. 1.2.1 MicrofractureTechnique While chondral defects (those limited to the cartilage layer) do not spontaneously heal due in part to the reasons mentioned above, wounds that penetrate the sub-chondral plate into the underlying bone space have much greater intrinsic healing capacity [18, 66]. Since bone is much more vascular than cartilage, wounds that penetrate into the bone space result in the formation of a fibrin clot within the lesion and introduce a supply of cells and growth factors that initiate a repair response. Although the native chondrocytes do not contribute substantially to the repair process [60], mesenchymal 10 stem cells derived from bone marrow can differentiate into chondrocytes or osteoblasts (bone-forming cells) as the new repair tissue is formed. The microfracture technique attempts to stimulate this spontaneous healing response by extending chondral lesions into the underlying bone space. This arthroscopic procedure introduces small holes in the subchondral bone plate by means of an awl, inducing bleeding from the bone space into the damaged cartilage [63]. Although positive results have been reported with use of the microfracture technique [61, 63], there are inherent disadvantages to the procedure. The spontaneous healing response to osteochondral defects generally results in repair with fibrocartilage, which is not durable over the long-term under physiological loading conditions [5, 48, 60, 64]. Additionally, the microfracture technique compromises the mechanical integrity of the subchondral plate, albeit to a lesser extent than procedures employing drills or pins (e.g. Pridie drilling and abrasion chondroplasty) [27, 55]. 1.2.2 Tissue and Cell Transplantation Other clinical approaches to treating chondral defects involve transplantation of tissue or cells from a relatively low-weight-bearing region of the cartilage in the same or other joint. In the mosaicplasty procedure, osteochondral plugs are removed from the edge of the patellar groove or proximal to the intercondylar notch and transplanted into the cartilage defect [17, 55]. In the cell-based procedure, referred to as autologous chondrocyte implantation (ACI), harvested tissue is enzymatically digested to obtain the cartilage cells, which are then expanded in culture and injected into the defect underneath a periosteal graft [8]. One readily apparent drawback to these transplantation procedures is donor site morbidity. Although the harvest site may temporarily fill with fibrocartilaginous repair tissue, it is expected to degenerate in the long-term [27]. Furthermore, the harvest procedure has been shown to adversely affect the mechanical properties of surrounding tissue [39]. In addition to donor site morbidity, animal investigations utilizing the mosaicplasty technique have revealed short-term degeneration of both the transplanted tissue and surrounding host cartilage [2, 37]. Additional difficulties encountered with transplantation of chondrocytes include in vitro expansion of the cells [46, 56], retention 11 of the transplanted cells within the defect [6, 7], integration of newly synthesized tissue with host tissue [6, 8], and damage to host tissue caused by the suturing procedure [7]. 1.3 Prospects for Tissue Engineering Tissue engineering employs the three major components of tissues - cells, extracellular matrix, and soluble regulators - or substitutes thereof, either alone or in combination, in efforts to restore the form and function of damaged tissue [36]. While tissue-engineering approaches utilizing each of these components alone (such as ACI) have yet to succeed in producing the desired results of regenerated articular cartilage, methods employing combinations of these components (viz. cells and ECM analogs) have been successful in regeneration of other tissues (see Yannas [68] for review) and have shown promise for articular cartilage [5]. Many different types of matrices have been investigated, such as fibrin, collagen, polyglycolic or polylactic acid, agarose, alginate, and synthetic polymers such as Teflon and Dacron [27]. Cells that have been investigated for use with these matrices, in addition to autologous and allogenic chondrocytes, include chondroprogenitor cells from sources such as periosteum and perichondrium [40], and bone marrow [9, 34]. Another source of cells that has drawn interest of late is the synovium. Synovial cells have recently been implicated in various cartilage repair procedures in vivo [26, 28], and their chondrogenic potential has been demonstrated in vitro [13, 54]. Synovium is both spontaneously regenerative [31] - thus eliminating the problem of donor site morbidity - and easily accessible during explorative or therapeutic arthroscopy. It is thus anticipated that this tissue may be used as a source of donor cells for articular cartilage tissue engineering. In designing tissue-engineered implants it is necessary to understand various aspects of the cell-matrix interaction. For example, a variety of musculoskeletal connective tissue cells, when cultured in collagen scaffolds, have been shown to contract by means of their expression of the contractile actin isoform, cc-smooth muscle actin (SMA) [62]. Such cellular contraction results in architectural deformation of the scaffold, which could contribute to failure of the engineered construct when implanted in vivo. 12 The purpose of this study was to lay the groundwork for development of a tissueengineered implant, composed of cells from the synovial membrane seeded in a collagenglycosaminoglycan (GAG) scaffold, to facilitate regeneration of articular cartilage. Toward this goal, the SMA expression of the synovial cells and the related cell-mediated contraction of the constructs were examined, as well as cellular proliferation and chondrogenesis within the scaffolds. 1.4 1.4.1 Review of Synovium, Collagen-GAG Scaffolds, and SMA-Enabled Contraction Synovium Structure and Function Synovium is a thin membrane of connective tissue found on the innermost lining of the joint capsule in diarthroidal joints (see Hung [24]for review). It is composed of an intimal layer of epithelial cells and a sub-intimal stroma composed primarily of fibrous, adipose, and areolar connective tissue. Unlike most connective tissues, synovium lacks a basement membrane separating the epithelial and stromal regions. The synovial intima is a layer of cells usually 2-3 cells deep comprising two cell types referred to as type A and type B synoviocytes. The type A cells are phenotypically similar to macrophages, being highly phagocytic and staining immunohistochemically for a number of macrophage markers. The type B cells resemble fibroblasts ultrastructurally, but differ from sub-intimal fibroblasts in that they stain with a specific monoclonal antibody (MAB 67) and are marked by a high level of uridine diphosphoglucose dehydrogenase (UDPGD) activity, a precursor to hyaluronan synthesis. The synovium is responsible for two primary functions. The first is control of transsynovial diffusion, which supplies nutrients to the avascular meniscal and cartilaginous tissues and regulates intra-articular pressures. The second function is synthesis of molecular components of synovial fluid, including hyaluronan and lubricin. The chondrogenic potential of synovial cells under certain pathological conditions has been recognized for some time. In synovial chondromatosis, cartilage-like nodules are found in various parts of the synovial membrane [45]. Also, a pannus of granulation tissue in which cells appear to display both synovial and chondrocyte-like characteristics is often found in cases of rheumatoid arthritis [3]. In addition to pathologic conditions, 13 synovial cells have recently been shown to display a chondrocytic phenotype when exposed to certain growth factors both in vivo and in vitro. 1.4.2 Collagen-GAG matrices The porous collagen-glycosaminoglycan scaffolds employed in this study were first developed by for dermal tissue engineering [69]. Recently, these scaffolds, composed of type 1 collagen and chondroitin-6-sulfate, have demonstrated promise in facilitating regeneration of peripheral nerves [10, 11], conjunctiva [23], intervertebral disk annulus [19], and articular cartilage [5]. These matrices have been employed as an analog of extracellular matrix in investigations of a variety of connective tissue cellmatrix interactions, including SMA-enabled contraction [62]. 1.4.3 SMA-Enabled Contraction Actin is a cytoskeletal protein associated with cellular shape, migration, and contraction. In humans there are six isoforms of actin, each encoded by a different gene. Four of these actin isoforms - y-smooth (enteric) muscle, a-skeletal muscle, a-cardiac muscle, and a-smooth (vascular) muscle (SMA) - are critical components of the contractile apparatus of their respective muscle cell types. It was initially thought that SMA was only expressed by vascular smooth muscle cells, but later was found in fibroblasts responsible for the contraction observed during healing of skin wounds [20, 41]. More recently, studies have demonstrated that some musculoskeletal connective tissue cells - including chondrocytes [33, 43], fibrochondrocytes [49], ligament fibroblasts [52], and osteoblasts [14, 47] - can express SMA and can contract scaffolds in which they are grown. While the physiological roles of SMA-enabled contraction of these cells have yet to be established, cell-mediated contraction of scaffolds can alter the pore diameter of the matrix and distort its overall shape, and thus needs to be addressed in the design of tissue-engineered implants. 14 2 SPECIFIC AIMS AND WORKING HYPOTHESES In order to lay the groundwork for future investigations employing synovial cell- seeded collagen-GAG matrices for regeneration of articular cartilage, the specific aims of this thesis were as follows: 1. To investigate the expression of SMA by synovial cells in vivo. 2. To evaluate the in vitro expression of SMA by synovial cells with passage in monolayer culture. 3. To evaluate the contraction of the collage-GAG matrices by the synovial cells. 4. To examine the chondrogenic potential of the synovial cells when cultured in the collagen-GAG matrices. The working hypotheses of the investigations were: 1. SMA-expressing cells are present in normal, adult, canine synovium. 2. The SMA content of the cells increases with passage number in monolayer culture. 3. The synovial cells will contract the collagen-GAG matrices into which they were seeded. 4. The synovial cells will express a chondrogenic phenotype when cultured in collagen-GAG matrices in the presence of a chondrogenic medium. 15 16 3 3.1 MATERIALS AND METHODS Synovial Cell Isolation and Culture Specimens of synovial membrane were obtained aseptically from the knee joints of 6 dogs immediately postmortem and washed extensively in phosphate buffered saline (PBS; Life Technologies, Grand Island, NY) supplemented with antibiotic/antimycotic solution (100 U/ml penicillin, 100 tg/ml streptomycin, 0.25 tg/ml amphotericin B, Life Technologies). Representative samples from 5 of the animals were allocated for histological and immunohistochemical evaluation. Specimens were finely diced and placed in 25 cm 2 culture flasks in complete medium consisting of Dulbecco's Modified Eagle's Medium/Nutrient Mixture F12 (DMEM/F12, Life Technologies), 10% fetal bovine serum (FBS; Hyclone Laboratories, Logan, UT), 2% ascorbic acid phosphate (Sigma Chemicals, St. Louis, MO), and 1% antibiotics. Culture conditions were maintained at 37*C in an atmosphere of 5% CO 2 and 95% humidity. Medium was first changed after 4 to 5 days in order to allow the tissue to attach to the culture flask, then was changed every 2 to 3 days. Once cells growing out of the tissue pieces reached a confluent layer covering approximately 50% of the flask surface (i.e., passage 1), tissue pieces were removed and cells were released by treatment with EDTA-Trypsin (Sigma) and sub-cultured in 75 cm 2 flasks for seven subsequent passages (P2-P8). Seeding density at each passage was 1.33x 104 cells/cm 2. The time required for the cells to reach confluence ranged from 6 to 8 days. Growth kinetics were determined by the following formula: Population doublings per day = ln(N/No) * t- 1, where t is the time period, N is the cell number at the end of the time period, t, and No is the cell number at the beginning of the time period [42]. During the second passage, a portion of the cells from each animal was suspended in complete medium containing 10% dimethylsulfoxide (DMSO, Sigma) and frozen at -70'C for up to 6 months prior to use in the chondrogenic differentiation assays (see section 3.3). 17 3.2 3.2.1 Three-Dimensional Cell Culture Using Collagen-GAG Scaffolds Matrixfabrication Porous type I collagen-GAG scaffolds were fabricated using a procedure previously described [69] (See Appendix A for detailed protocols). A co-precipitate of type I collagen from bovine tendon (Integra Life Sciences, Plainsboro, NJ) and chondroitin-6-sulfate from shark cartilage (Sigma) was freeze-dried to produce sheets approximately 3 mm in thickness. After dehydrothermal cross-linking for 24 hours, 9 mm diameter samples were cut from the sheets. Prior work has shown these matrices to have a porosity of approximately 87% and a mean pore diameter of 83 pm [53]. 3.2.2 Cell-Seeding Prior to seeding, matrix samples were soaked in PBS for 1 hour, transferred to complete medium for 10 minutes, and dried briefly on sterile filter paper. Each matrix was seeded with P4 cells by pipetting a suspension of 2x 106 cells in 50 pl medium onto the surfaces of the matrix (25 pl per side), resulting in a density of approximately lx 104 cells/mm 3. Cell-seeded matrices and non-seeded controls were cultured in 20 mm diameter wells of 12-well plates coated with 1.5 ml of agarose (2% w/v) to prevent the cells from attaching to the polystyrene surface. Complete medium (0.5 ml) was added to each well 2 hours post-seeding, followed by another 1.0 ml 12-16 hours later. Media were changed every other day. 3.2.3 Matrix ContractionMeasurements The diameters of the cell-seeded matrices as well as non-seeded controls cultured in parallel under identical conditions were measured by visual inspection every other day using circular templates ranging from 1 mm to 10 mm diameter in 0.5 mm increments. The change in diameter of the matrices was expressed as a percentage reduction in the diameter of the scaffolds based on the starting diameter of the matrices prior to hydration on the day of seeding. The contraction of the non-seeded scaffolds was subtracted from the contraction of the cell-seeded samples to yield a measure of "cell-mediated contraction." Cultures were terminated after 1, 7, 14 and 28 days post-seeding for histology and analysis of DNA content. 18 3.3 Chondrogenic Differentiation culture A micro-pellet culture system previously described [30] was employed to investigate the chondrogenic potential of the synovial cells. Cells frozen at -70'C were thawed, rinsed of DMSO by centrifugation, and expanded through two passages in monolayer culture. Aliquots of 2.5 x 105 cells (P4) were gently centrifuged in 15 ml centrifuge tubes to form free-floating pellets and incubated in either: 1) an FBSsupplemented medium made up of DMEM/F 12 with 10% FBS, 10 ng/ml TGF-p 1, 50 pg/ml ascorbic acid phosphate, and IO0nM dexamethasone; or 2) a defined medium consisting of DMEM (high-glucose with 110 tg/ml sodium pyruvate, Sigma), "ITS+ Premix" (BD Biosciences) - insulin (6.25 ptg/ml), transferrin (6.25 ptg/ml), selenous acid (6.25 pg/ml) linoleic acid (5.35 pg/ml), and bovine serum albumin (1.25 pg/ml) - and 10 ng/ml TGF-$P1, 50 pg/ml ascorbic acid phosphate, and lOOnM dexamethasone. Articular chondrocytes cultured in parallel under identical conditions served as a control. In order to investigate the chondrogenic differentiation potential of the synovial cells in the collagen-GAG matrix culture system, P4 cells were seeded into matrices and cultured in the defined or FBS-supplemented media (described above). After three weeks, cultures were fixed in formalin. Paraffin-embedded sections were evaluated for chondrogenic differentiation by immunohistochemical detection of type II collagen. 3.4 Western Blot Analysis for SMA At each passage (P1-P8) of the synovial cells in monolayer culture, an aliquot of 1-2 x 106 cells was removed for Western blot analysis of SMA content. Cytoplasmic proteins were extracted from the cells using a lysing buffer (1% sodium dodecyl sulfate, 1 mM sodium orthovanadate, 1 mM phenylmethanesulfonyl fluoride, and 10% glycerol). The cell lysates were agitated at 4'C for 10 minutes, then centrifuged for 20 minutes at 14,000 rpm at 4*C. The supernatant containing the protein was removed and stored at -20*C until evaluation. The concentrations of extracted proteins relative to a BSA standard were determined by a modification of the Bradford method [4]. Proteins were mixed with BioRad Protein Assay Dye (Bio-Rad Laboratories, Hercules, CA) and the optical density measured at a wavelength of 595 nm using a LKB Biochrom Ultraspec 4050 19 spectrophotometer. Cell extracts containing 5 pg of total protein were diluted with sample buffer and dH 20 to a total of 48 pil, resolved in a 12% polyacrylamide gel in a mini-gel apparatus (Bio-Rad Mini-Protean II, Bio-Rad) for 90 minutes at 90 V, then transferred to PVDF membranes for 60 minutes at 100 V. Extracts from human aorta smooth muscle cells containing 5 tg of protein served as positive controls. After transfer, blotted membranes were placed in 5% dry milk (Bio-Rad) overnight to block non-specific binding of antibodies. Immunoprobing of the blotted proteins was achieved by incubation with the primary antibody (Clone 1A4, Monoclonal Anti-SMA, Sigma) for 2 hours. Bound protein was detected by incubation with a peroxidase-conjugated secondary antibody (goat anti-mouse IgG, Sigma) for one hour followed by reaction with a luminol-based chemiluminescent reagent (LumiGlo, Cell Signaling Technology, Beverly, MA). Film (Kodak Scientific Imaging Film) exposed by contact with the blotted membranes was developed and digitized for densitometric analysis using Scion Image software (Scion Corp., Frederick, MD). The densities of the bands corresponding to SMA (molecular weight of 42 kDa) for the experimental samples were each divided by the density of the band for the SMC positive control in the same blot. This provided a relative measure of SMA content as a percentage of the SMC positive control and allowed for comparison among blots. 3.5 DNA analysis Cell-seeded scaffolds and non-seeded control matrices allocated for analysis of DNA content were washed in PBS and stored at -20*C until assayed. After lyophilization, the matrices were digested overnight in a papain buffer (6 pg papain in 0.1 M sodium phosphate, 5 mM Na2 EDTA, and 10 mM cysteine-HCL) at 60*C. DNA content in the digests was determined by the Hoechst dye method [35] using calf thymus DNA as a standard. One hundred pl of digest was combined with 2 ml of Hoechst dye working solution (#33258, Polyscience Inc., Northampton, UK) and evaluated fluorometrically (TKO 100 Fluorometer, Hoeffer Scientific Instruments, San Francisco, CA). The background fluorescence of the matrix was accounted for by subtracting the mean value obtained for non-seeded controls from the value of each cell-seeded construct at each time point. 20 3.6 Histology and Immunohistochemistry Tissue samples and cell-seeded matrices were fixed in 10% formalin, processed, and embedded in paraffin for microtomy. Seven pm thick sections were deparaffinized and stained with hematoxylin and eosin using standard histological techniques (see Appendix D). Sections allocated for immunohistochemical analysis were stained with antibodies to SMA or type II collagen (detailed protocols in Appendix D). For SMA analysis, deparaffinized and rehydrated sections were digested in 0.1% trypsin for 1 hour, followed by quenching of endogenous peroxidase with 3% hydrogen peroxide. Non-specific binding was blocked by incubation with 30% goat serum (Sigma). Samples were then incubated with primary antibody (Clone IA4, Monoclonal Anti-SMA, Sigma) for 2 hours at room temperature. For negative controls, one section on each slide was incubated with non-immunologic mouse serum (Sigma) diluted to the same protein concentration, instead of the primary antibody. Sections were then incubated with a biotinylated secondary antibody (goat anti-mouse IgG, Sigma), followed by application of affinitypurified avidin (Sigma). Labeling was developed using an aminoethyl carbazole (AEC) chromagen kit (Zymed Laboratories, San Francisco, CA). Counterstaining was performed using Mayer's hematoxylin (Sigma). For analysis of type II collagen, deparaffinized and rehydrated sections were digested for 1 hour in 0.1% protease XIV, followed by blocking of non-specific binding with 5% horse serum. Primary antibody (CIIC 1, mouse anit-chick type II collagen monoclonal antibody, Developmental Studies Hybridoma Bank, Iowa City, IA) was applied for 1 hour. Negative controls were incubated with mouse IgG (Zymed) diluted to the same protein concentration, instead of the primary antibody. A biotinylated secondary antibody (horse anti-mouse IgG, Sigma) was applied for 45 minutes, followed by quenching of endogenous peroxidase with 3% hydrogen peroxide. Labelling was detected with an avadin-biotin complex [21, 22] (ABC kit, Vectastain), Vector Laboratories, Burlingame, CA), and diamenobenzadine (DAB, Vector). Counterstaining was performed using Harris hematoxylin. 21 3.7 Statistical Analysis Statistical significance was determined by analysis of variance (ANOVA) with a significance criterion of p<0.0 5 using StatView (SAS Institute Inc., Cary, N.C.) Curvefitting was accomplished using Igor Pro4 (WaveMetrics Inc, Lake Oswego, OR). 22 4 4.1 RESULTS SMA Expression In Vivo Immunohistochemical staining revealed the presence of SMA in tissue from 4 out of 5 animals evaluated. Approximately 2-5% of cells in the intimal layer of these synovium samples stained positive for SMA (Fig. 1). No significant staining could be detected in cells from the sub-intimal tissue, with the exception of perivascular smooth muscle cells. The intensity of staining of cells in the intima that were SMA-positive was similar to that of the smooth muscle cells, which were used as positive internal controls. b Figure 1: Immunohistochemical staining for a-smooth muscle actin (SMA) in 2 samples of intact adult canine synovium (red chromogen, see arrows). Scale bars = 20 pim. Insets are negative controls. 23 24 Cell Growth and In Vitro Expression of SMA 4.2 Cell Growth in Monolayer Culture Cell growth rate in monolayer culture tended to decrease with passage number, with P8 cells growing at approximately half the rate of P3 cells (Fig. 2). ANOVA 4.2.1 revealed a significant effect of passage number on the number of population doublings per day (P < 0.02). 0.4 0.3 .2 0 0 IL 0 1 P2 P3 P4 P5 P6 P7 P8 Passage Number Figure 2: Change in cell growth rate in monolayer culture with passage number (n=2-7). Mean ± standard error of the mean (SEM) 4.2.2 Western Blot Analysis of SMA Content Western blot analysis detected SMA in P1 cells from each of the 6 animals, although the intensity of two samples was not strong enough for meaningful densitometric quantification. Blots of extracted protein from P2 to P8 cells revealed a regular increase in SMA content with passage number, reaching a level nearly equivalent to that found in the smooth muscle cell controls by the fourth passage (Fig. 3). By the seventh passage the SMA content of the synovial cells was elevated nearly 20-fold compared to P1 cells. One factor ANOVA revealed a significant effect of passage number (P<0.001) on SMA content of the cells. There was also a significant effect of 25 length of time in culture on SMA content of the cells (P<0.0002), but regression analysis showed that there was not a meaningful linear correlation (Fig.4; R2 = 0.46). 250 0 * 2000 150 0100 0 50 0 2 U, P1 P2 III P3 P5 P4 P6 P7 P8 Passage Number Figure 3: Increase in SMA content of synovial cells with passage number, expressed as a percentage of the smooth muscle cell positive controls. Mean SEM; P2-P5, n=6, P1 and P6-P8, n=3. 300 0 L- 1250 C 50 /0 &150 0 10 30 40 50 60 Time in Culture (Days) Figure 4: Scatter plot showing the SMA content of synovial cells with time in monolayer culture, and a linear regression analysis (R2 = 0.46). 26 4.3 Contraction of Collagen-GAG scaffolds DiameterMeasurements Non-seeded control scaffolds contracted to 71% of the original diameter after 28 days. In contrast, cell-seeded constructs contracted to 27% of the original diameter (4% of the original volume) over the same time period (Fig. 5). After only 1 week, cell- 4.3.1 seeded matrices were reduced in diameter by more than 50%. Two-factor ANOVA revealed a significant effect of whether the scaffolds were seeded with cells (P<0.0001) and time in culture (P<0.0001) on contraction. inn rr~ 80 E 0) 60 - 40 - %0 --s- Non-seeded controls 20 -o- Cell-Seeded 0 0 5 10 15 20 25 30 Time (Days) Figure 5: Decrease in diameter of non-seeded control matrices (n=5) and of scaffolds seeded with P4 cells (n=36). Sample numbers of cell-seeded scaffolds decreased to 24 and 12 after 7 and 14 days. Mean ±SEM. The cell-mediated contraction data were fit with a curve of form A(1 -e-th) having an asymptote (A) of 43% of the original scaffold diameter and a time constant (r) of 2.4 days (Fig. 6). 27 50 IL * 40 - .5 30 - Uo 20 - .L 0 E 0 (U (U (U ., Cell-mediated Contraction .5) 10- - Curve Fit 0 0 5 20 15 10 25 30 Time (Days) Figure 6: Cell-mediated contraction of the collagen-GAG matrices and exponential fit of the form A(l-exp(-t/t)) with asymptote, A = 43%, of the original scaffold diameter and time constant, t = 2.4 days. 4.3.2 DNA Content The DNA content of the constructs diminished throughout the 4-week culture period (Fig 7). One factor ANOVA showed a significant effect of time in culture on the DNA content of the constructs (P=0.01). The DNA value at one day reflected approximately 1.2x 106 cells (assuming an average DNA content of 7.7 pg DNA per cell, as has been reported for chondrocytes [32]), indicating that about 60% of the seeded cells were attached to the scaffold after this time period. 15000 12500 10000 r 7500 z a I T 5000 2500 01 7 14 28 Time in Culture (days) Figure 7: DNA content of the cell-seeded matrices. Mean ±SEM. 28 4.3.3 Histology of the Cell-Seeded Matrices Staining of cell-seeded matrices with hematoxylin and eosin revealed the distribution of the synovial cells throughout the collagen-GAG scaffolds with a generally greater cell density on the surface as compared to the interior. Constructs terminated 1 day post-seeding demonstrated an open porous network, with relatively uniform pore size of about 100 pm throughout (Fig. 8a). After 1 week a continuous cell layer could be seen forming on the periphery of the constructs that stained positive for SMA (Fig. 8b). Pores near the periphery of these scaffolds appeared compressed and were markedly smaller than those in the central region of the constructs. Compression and collapse of the pores was seen to progress toward the interior of the constructs until, after 4 weeks, there was virtually no porosity remaining in many of the samples (Fig. 8c). 4.4 Chondrogenic Differentiation of Synovial Cells Some cell pellets (Figs. 9a and 9b) and cell-seeded matrices (Figs. 9c-d) cultured in the defined differentiation medium stained positive immunohistochemically for type II collagen, but only in trace amounts. Several sections from these specimens did not reveal any positive staining. The extent of type II collagen staining was considerably less than that found in the articular cartilage tissue control (Fig. 9e) and in the chondrocyte micropellet control (Fig. 9f). No significant staining for type II collagen could be detected in any of the pellets or matrices cultured with the FBS-supplemented differentiation medium. 29 30 __ Figure 8: a) Histological micrograph of a synovial cell-seeded scaffold after 1 day in culture. Hematoxylin and eosin stain; scale bar = 250 gm. b) Immunohistochemical staining of SMA (red chromogen, arrows) in a synovial cell-seeded scaffold after 7 days in culture. Scale bar = 50 ptm. c) Synovial cell-seeded scaffold after 28 days in culture. Hematoxylin and eosin stain; scale bar = 250 ptm. 31 ____ - I 32 EF~~~fmWAkW ~ m N 3E Ib~l~m Figure 9: Immunohistochemical staining of type 11 collagen. Brownish chromogen (see arrows) indicates presence of type 11 collagen. (a) Synovial cell micro-pellet culture and (b) negative control. Scale bars = 20 pm. (c and d) Synovial cell-seeded matrix cultures. Scale bars =20 p m. (e) Canine articular cartilage and (f') chondrocyte micro-pellet culture used as positive controls. Scale bars = 200 p.. 33 34 5 DISCUSSION 5.1 SMA Expression In Vivo The immunohistochemical detection of SMA in specimens of intact synovium provides confirmation of the first hypothesis presented in Section 2. This is the first report of SMA expression in cells comprising the intimal layer of normal adult synovium in vivo. Previous studies have reported the expression of SMA in synovial cells under various pathologic conditions. Murray et al. identified SMA positive cells within the synovial layer formed during the retraction of ruptured anterior cruciate ligaments and postulated that the contraction of such cells contributed to the retraction of the ends of the ligament. [51]. SMA was also identified in vitro by immunohistochemistry in synovial cells from osteoarthritic and rheumatoid patients, though no significant staining of SMA was found in vivo [44]. That the number of cells expressing SMA in vivo varied among the samples examined in this study, with sections from one animal demonstrating no SMA positive cells, indicates a transient nature of the SMA expression, as has been suggested for fibroblasts [12, 58, 67]. Of particular interest is the result that in all of the samples, SMA expressing cells were localized only in the synovial intima and were not found in the fibroblast population of the sub-intimal tissue. These results warrant further investigation into the identity of the SMA-expressing cells as type A (macrophage-like) or type B (fibroblast-like) synoviocytes and the physiological role of SMA expression in synovial cells. 5.2 In Vitro Expression of SMA The finding that SMA expression by synovial cells increases with in vitro expansion provides confirmation of the second hypothesis of Section 2, and is consistent with data reported for chondrocytes [33], fibrochondrocytes [49], and osteoblasts [47]. Unlike data reported for chondrocytes [33], the relationship between SMA content of the synovial cells and the length of time in culture did not appear to be linear (Fig. 7). The presence of SMA in samples from intact synovium indicates that the contractility and SMA expression of the synovial cells was not solely due to the adoption of this phenotype in culture. However, the possibility of contribution to the in vitro results by 35 sub-intimal cells that did not exhibit SMA expression in vivo requires further consideration. 5.3 Chondrogenic Differentiation of Synovial Cells The detection of type II collagen in cultures of the synovial cells both in micro- pellet and collagen-GAG matrix culture systems provides confirmation of the fourth hypothesis of Section 2. The synthesis of type II collagen indicates their capacity to differentiate to a chondrocytic phenotype, as has been previously demonstrated [13, 54], and thus supports the use of these cells in cartilage tissue engineering. That no type II collagen could be detected in FBS-supplemented cultures may indicate the presence of a factor in the serum that inhibits chondrogenic differentiation of the cells. Similar results have been demonstrated for chondrocytes. Following dedifferentiation during monolayer culture, chondrocytes exhibit a greater extent of re-differentiation to a chondrocytic phenotype when cultured in chemically defined media than in the presence of serum [29]. That only trace amounts of type II collagen were detected in the cultures of synovial cells with defined media raises questions of the conditions that favor the differentiation of these cells to chondrocytes, and the degree to which such conditions exist in cartilage defects in vivo. 5.4 Contraction of Collagen-GAG scaffolds Results of the matrix contraction assay provide confirmation of the third hypothesis presented in Section 2. The reduction in diameter of non-seeded control matrices may have been due to the mild plasticizing effect of water on collagen, as has been reported [65]. While this effect was partly responsible for the decrease in diameter of cell-seeded scaffolds, contraction of over 40% of the original diameter can be attributed to the presence of the synovial cells. Recent studies have demonstrated the contractility of a variety of connective tissue cell types [33, 38, 47, 49, 59], including the progenitor mesenchymal stem cell [9, 34]. It may thus have been expected that cells of synovial origin are also capable of displaying a contractile phenotype. 36 Cell-mediated contraction leveled off after about 3 weeks (Fig. 6), likely due to the fact that virtually all of the porosity of the scaffold had been lost (Fig. 8c). Additionally, the DNA content of the cell-seeded matrices decreased throughout the culture period, indicating cell loss that may have been due to compression of the pores in the contracted scaffolds. Histology revealed compression of the pores on the surface of the scaffolds as early as seven days post-seeding, with collapse of the pores proceeding from the surface to the central regions of the scaffolds throughout the 4-week culture period. Such architectural deformation of engineered constructs caused by cellular contraction could contribute to failure when implanted in vivo by hindering the migration of the cells within the construct and preventing integration with surrounding host tissue. Although the molecular mechanisms underlying contraction of SMA-expressing non-muscle cells are not yet known, two investigations have indicated a causal relationship between SMA expression and cell contractility [33, 70]. The results of the current investigation may thus be of value in informing future efforts to expand synovial cells in vitro prior to implantation for cell-based therapies or tissue engineering procedures for cartilage repair, as well as efforts to manage cell-mediated contraction of engineered constructs and reparative tissue by use of regulators to control SMA expression. 37 38 6 CONCLUSIONS Following are conclusions related to the specific aims and working hypotheses of the study described in section 2: 1. Immunohistochemistry revealed SMA-expressing cells in the intimal cell layer of synovial membranes from 4 out of 5 of the animals investigated. No staining of SMA was detected in sub-intimal tissue. 2. SMA content of the synovial cells increased with passage number in monolayer culture. 3. Synovial cells contract collagen-GAG matrices into which they are seeded, causing collapse of the porous structure and loss of volume. 4. Synovial cells exhibit chondrogenic potential indicated by synthesis of type II collagen, albeit in trace amounts, when cultured as micro-pellets or in collagenGAG scaffold in the presence of a chemically defined chondrogenic medium. 39 40 7 LIMITATIONS AND FUTURE WORK There are several limitations to the current study that should be addressed in future work. First, tissues and cells used in the present study were from a single species (canine), and should only be considered directly applicable to this species. Future studies evaluating the SMA expression and contractility of human synovial cells will be beneficial. It is reasonable to expect results similar to the present study to be found in human synovial cells because findings of SMA expression and contractility in studies of in other human musculoskeletal connective tissue cells have paralleled findings in canine studies. Immunohistochemical staining of synovial tissue revealed SMA expressing cells in the intimal layer of the synovium, but not in cells from the sub-intimal tissue. It was assumed in this study that the increase in SMA content of cells with passage number in monolayer primarily reflected an increase in SMA expression by cells that expressed SMA in vivo. However, it is possible that cells from the sub-intimal tissue that did not express SMA in vivo may have grown out of the tissue and adopted an SMA-expressing phenotype, thus influencing the in vitro results. Future studies employing intimal cells and sub-intimal cells separately may allow for confirmation or rejection of this possibility. Such studies may have to await the development of markers to easily distinguish intimal cells from sub-intimal cells in vitro. The method of cell-isolation (outgrowth from tissue explants) chosen for this study did not allow quantitative evaluation of the SMA content of the cells prior to their expansion in monolayer culture (i.e. a PO time point). Additional studies employing alternative methods of cell isolation, such as enzymatic digestion, would allow opportunity for quantitative evaluation of SMA in freshly isolated cells and provide additional insight concerning the extent to which this phenotype is induced by in vitro culture of the cells. Assays of cell-mediated contraction and chondrogenic potential in this study employed cells that had been cultured in monolayer through Passage 4. Further studies should be conducted employing cells from a variety of passage numbers, as well as cells freshly isolated from explanted tissue. One would expect such studies to reveal cellular contractility increasing with passage number of the cells, consistent with the increase in 41 SMA demonstrated in the current study. Similar results have been demonstrated in chondrocytes [33]. Since increased SMA expression and related cellular contractility caused by prolonged in vitro expansion of the cells can be problematic in the context of a cell-seeded implant, it may be advantageous to employ cells from the earliest time point possible. As such, an arthroscopic procedure is anticipated in which cells are isolated from the synovium, seeded in a matrix, and implanted in a cartilage defect during the same surgical procedure. Methods of mechanically isolating the cells by means of an arthroscopic shaver, and instruments for intra-operatively seeding the cells in a matrix are currently under investigation. The present study examined several cell-matrix interactions important to the development of a synovial cell/collagen-GAG implant for cartilage regeneration. Future studies in the design of such an implant may examine effects of collagen type (i.e., type I versus type II collagen), methods of cross-linking the matrix to increase resistance to contraction, and the use of soluble regulators to control SMA expression and contraction and to promote chondrogenesis in the synovial cells. 42 REFERENCES 1. "Osteoarthritis", brochure from the Arthritis Foundation, 2002. 2. Aeschlimann, D., et al. "Repair of cartilage defects with autogenous osteochondral transplants (mosaicplasty) in a sheep model". in 46th Annual Meeting, Orthopaedic Research Society. 2000. Orlando. 3. Allard, S.A., R.N. Maini, and K.D. Muirden, "Cells and matrix expressing cartilage components in fibroblastic tissue in rheumatoid pannus". Scandinavian Journal of Rheumatology - Supplement, 1988. 76: p. 125-9. 4. Bradford, M.M., "A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding". Analytical Biochemistry, 1976. 72: p. 248-54. 5. Breinan, H.A., et al., "Healing of canine articular cartilage defects treated with microfracture, a type-Il collagen matrix, or cultured autologous chondrocytes". Journal of Orthopaedic Research, 2000. 18(5): p. 781-9. 6. Breinan, H.A., et al., "Histological Evaluation of the Course of Healing of Canine Articular Cartilage Defects Treated with Cultured Autologous Chondrocytes". Tissue Engineering, 1998. 4(1): p. 101-114. 7. Breinan, H.A., et al., "Effect of cultured autologous chondrocytes on repair of chondral defects in a canine model". Journal of Bone & Joint Surgery, 1997. 79(10): p. 1439-51. 8. Brittberg, M., et al., "Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. [see comments]". New England Journal of Medicine, 1994. 331(14): p. 889-95. 9. Cai, D., et al., "Lapine and canine bone marrow stromal cells contain smooth muscle actin and contract a collagen-glycosaminoglycan matrix". Tissue Engineering, 2001. 7(6): p. 829-41. 10. Chamberlain, L.J., et al., "Collagen-GAG substrate enhances the quality of nerve regeneration through collagen tubes up to level of autograft". Experimental Neurology, 1998. 154(2): p. 315-29. 11. Chamberlain, L.J., et al., "Near-terminus axonal structure and function following rat sciatic nerve regeneration through a collagen-GAG matrix in a ten-millimeter gap". Journal of Neuroscience Research, 2000. 60(5): p. 666-77. 12. Darby, I., 0. Skalli, and G. Gabbiani, "Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing". Laboratory Investigation, 1990. 63(1): p. 21-9. 13. De Bari, C., et al., "Multipotent mesenchymal stem cells from adult human synovial membrane". Arthritis & Rheumatism, 2001. 44(8): p. 1928-42. 14. Goldstein, A.S., et al., "Effect of osteoblastic culture conditions on the structure of poly(DL-lactic-co-glycolic acid) foam scaffolds". Tissue Engineering, 1999. 5(5): p. 421-34. 43 15. Grande, D.A., et al., "The repair of experimentally produced defects in rabbit articular cartilage by autologous chondrocyte transplantation". Journal of Orthopaedic Research, 1989. 7(2): p. 208-18. 16. Grande, D.A., I.J. Singh, and J. Pugh, "Healing of experimentally produced lesions in articular cartilage following chondrocyte transplantation". Anatomical Record, 1987. 218(2): p. 142-8. 17. Hangody, L., et al., "Arthroscopic autogenous osteochondral mosaicplasty for the treatment of femoral condylar articular defects. A preliminary report". Knee Surgery, Sports Traumatology, Arthroscopy, 1997. 5(4): p. 262-7. 18. Hanie, E.A., et al., "Healing of full-thickness cartilage compared with fullthickness cartilage and subchondral bone defects in the equine third carpal bone". Equine Veterinary Journal, 1992. 24(5): p. 382-6. 19. Hastreiter, D., "A collagen-GAG matrix for the growth of intervertebral disc tissue". 2002, Massachusettes Institute of Technology: Cambridge. 20. Hirschel, B.J., et al., "Fibroblasts of granulation tissue: immunofluorescent staining with antismooth muscle serum". Proceedings of the Society for Experimental Biology & Medicine, 1971. 138(2): p. 466-9. 21. Hsu, S.M., L. Raine, and H. Fanger, "A comparative study of the peroxidaseantiperoxidase method and an avidin-biotin complex method for studying polypeptide hormones with radioimmunoassay antibodies". American Journal of Clinical Pathology, 1981. 75(5): p. 734-8. 22. Hsu, S.M., L. Raine, and H. Fanger, "Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures". Journal of Histochemistry & Cytochemistry, 1981. 29(4): p. 577-80. 23. Hsu, W.C., et al., "Inhibition of conjunctival scarring and contraction by a porous collagen-glycosaminoglycan implant". Investigative Ophthalmology & Visual Science, 2000. 41(9): p. 2404-11. 24. Hung, G.L. and C. Evans, "Synovium", in Knee Surgery, F. Fu, C.D. Hamer, and K.G. Vince, Editors. 1994, Williams and Williams: Baltimore. p. 141-153. 25. Hunter, W., "On the structure and diseases of articulating cartilage". Philos Trans R Soc Lond, 1743. 42B: p. 514-521. 26. Hunziker, E.B., "Growth-factor-induced healing of partial-thickness defects in adult articular cartilage". Osteoarthritis & Cartilage, 2001. 9(1): p. 22-32. 27. Hunziker, E.B., "Articular cartilage repair: basic science and clinical progress". Osteoarthritis & Cartilage, 2002. 10(6): p. 432-463. 28. Hunziker, E.B. and L.C. Rosenberg, "Repair of partial-thickness defects in articular cartilage: cell recruitment from the synovial membrane". Journal of Bone & Joint Surgery - American Volume, 1996. 78(5): p. 721-33. 44 29. Jakob, M., et al., "Specific growth factors during the expansion and redifferentiation of adult human articular chondrocytes enhance chondrogenesis and cartilaginous tissue formation in vitro". Journal of Cellular Biochemistry, 2001. 81(2): p. 368-77. 30. Johnstone, B., et al., "In vitro chondrogenesis of bone marrow-derived mesenchymal progenitor cells". Experimental Cell Research, 1998. 238(1): p. 265-72. 31. Key, J.A., "The reformation of synovial membrane in the knees of rabbits after synovectomy". The Journal of Orthopedic Research, 1927: p. 793-813. 32. Kim, Y.J., et al., "Fluorometric assay of DNA in cartilage explants using Hoechst 33258". Analytical Biochemistry, 1988. 174(1): p. 168-76. 33. Kinner, B. and M. Spector, "Smooth muscle actin expression by human articular chondrocytes and their contraction of a collagen-glycosaminoglycan matrix in vitro". Journal of Orthopaedic Research, 2001. 19(2): p. 233-41. 34. Kinner, B., J.M. Zaleskas, and M. Spector, "Regulation of smooth muscle actin expression and contraction in adult human mesenchymal stem cells". Experimental Cell Research, 2002. 278(1): p. 72-83. 35. Labarca, C. and K. Paigen, "A simple, rapid, and sensitive DNA assay procedure". Analytical Biochemistry, 1980. 102(2): p. 344-52. 36. Langer, R. and J.P. Vacanti, "Tissue engineering". Science, 1993. 260(5110): p. 920-6. 37. Laurencin, C.T., et al., "Tissue engineering: orthopedic applications". Annual Review of Biomedical Engineering, 1999. 1: p. 19-46. 38. Lee, C.R., et al., "Articular cartilage chondrocytes in type I and type II collagenGAG matrices exhibit contractile behavior in vitro". Tissue Engineering, 2000. 6(5): p. 555-65. 39. Lee, C.R., et al., "Effects of harvest and selected cartilage repair procedures on the physical and biochemical properties of articular cartilage in the canine knee". Journal of Orthopaedic Research, 2000. 18(5): p. 790-9. 40. Lee, C.R. and M. Spector, "Status of articular cartilage tissue engineering". Current Opinion in Orthopaedics, 1998. 9: p. 88-93. 41. Majno, G., et al., "Contraction of granulation tissue in vitro: similarity to smooth muscle". Science, 1971. 173(996): p. 548-50. 42. Martin, B., Tissue Culture Techniques: An Introduction. 1994, Boston: Birkhauser. 43. Martin, I., et al., "Mammalian chondrocytes expanded in the presence of fibroblast growth factor 2 maintain the ability to differentiate and regenerate three-dimensional cartilaginous tissue". Experimental Cell Research, 1999. 253(2): p. 681-8. 45 44. Mattey, D.L., et al., "Transforming growth factor beta 1 and interleukin 4 induced alpha smooth muscle actin expression and myofibroblast-like differentiation in human synovial fibroblasts in vitro: modulation by basic fibroblast growth factor". Annals of the Rheumatic Diseases, 1997. 56(7): p. 426-3 1. 45. Maurice, H., M. Crone, and I. Watt, "Synovial chondromatosis". Journal of Bone & Joint Surgery - British Volume, 1988. 70(5): p. 807-11. 46. Mayhew, T.A., et al., "Validation of a quality assurance program for autologous cultured chondrocyte implantation". Tissue Engineering, 1998. 4(3): p. 325-34. 47. Menard, C., S. Mitchell, and M. Spector, "Contractile behavior of smooth muscle actin-containing osteoblasts in collagen-GAG matrices in vitro: implant-related cell contraction". Biomaterials, 2000. 21(18): p. 1867-77. 48. Minas, T. and S. Nehrer, "Current concepts in the treatment of articular cartilage defects". Orthopedics, 1997. 20(6): p. 525-38. 49. Mueller, S.M., et al., "alpha-smooth muscle actin and contractile behavior of bovine meniscus cells seeded in type I and type II collagen-GAG matrices". Journal of Biomedical Materials Research, 1999. 45(3): p. 157-66. 50. Muir, H., "The chondrocyte, architect of cartilage. Biomechanics, structure, function and molecular biology of cartilage matrix macromolecules". Bioessays, 1995. 17(12): p. 1039-48. 51. Murray, M.M., et al., "Histological changes in the human anterior cruciate ligament after rupture". Journal of Bone & Joint Surgery, 2000. 82-A(10): p. 1387-97. 52. Murray, M.M., S.D. Martin, and M. Spector, "Migration of cells from human anterior cruciate ligament explants into collagen-glycosaminoglycan scaffolds". Journal of Orthopaedic Research, 2000. 18(4): p. 557-64. 53. Nehrer, S., et al., "Matrix collagen type and pore size influence behaviour of seeded canine chondrocytes". Biomaterials, 1997. 18(11): p. 769-76. 54. Nishimura, K., et al., "Chondroprogenitor cells of synovial tissue". Arthritis & Rheumatism, 1999. 42(12): p. 2631-7. 55. O'Driscoll, S.W., "The healing and regeneration of articular cartilage". Journal of Bone & Joint Surgery, 1998. 80(12): p. 1795-812. 56. Peterson, L., et al., "Two- to 9-year outcome after autologous chondrocyte transplantation of the knee". Clinical Orthopaedics & Related Research, 2000(374): p. 212-34. 57. Rand, J.A. and D.M. Ilstrup, "Survivorship analysis of total knee arthroplasty. Cumulative rates of survival of 9200 total knee arthroplasties. [see comments.]". Journal of Bone & Joint Surgery, 1991. 73(3): p. 397-409. 58. Sappino, A.P., W. Schurch, and G. Gabbiani, "Differentiation repertoire of fibroblastic cells: expression of cytoskeletal proteins as marker of phenotypic modulations". Laboratory Investigation, 1990. 63(2): p. 144-61. 46 59. Schneider, T.O., et al., "Expression of alpha-smooth muscle actin in canine intervertebral disc cells in situ and in collagen-glycosaminoglycan matrices in vitro". Journal of Orthopaedic Research, 1999. 17(2): p. 192-9. 60. Shapiro, F., S. Koide, and M.J. Glimcher, "Cell origin and differentiation in the repair of full-thickness defects of articular cartilage". Journal of Bone & Joint Surgery, 1993. 75(4): p. 532-53. 61. Sledge, S.L., "Microfracture techniques in the treatment of osteochondral injuries". Clinics in Sports Medicine, 2001. 20(2): p. 365-77. Spector, M., "Musculoskeletal connective tissue cells with muscle: expression of muscle actin in and contraction of fibroblasts, chondrocytes, and osteoblasts". Wound Repair Regeneration, 2001. 9(1): p. 11-8. 62. 63. Steadman, J.R., W.G. Rodkey, and J.J. Rodrigo, "Microfracture: surgical technique and rehabilitation to treat chondral defects". Clinical Orthopaedics & Related Research, 2001(391 Suppl): p. S362-9. 64. Temenoff, J.S. and A.G. Mikos, "Review: tissue engineering for regeneration of articular cartilage". Biomaterials, 2000. 21(5): p. 431-40. 65. Torres, D.S., et al., "Tendon cell contraction of collagen-GAG matrices in vitro: effect of cross-linking". Biomaterials, 2000. 21(15): p. 1607-19. 66. Vachon, A., et al., "Evaluation of the repair process of cartilage defects of the equine third carpal bone with and without subchondral bone perforation". American Journal of Veterinary Research, 1986. 47(12): p. 2637-45. 67. Woodcock-Mitchell, J., et al., "Alpha-smooth muscle actin is transiently expressed in embryonic rat cardiac and skeletal muscles". Differentiation, 1988. 39(3): p. 161-6. 68. Yannas, I.V., Tissue and Organ Regeneration in Adults. 2001: Springer Verlag. 383. 69. Yannas, I.V., et al., "Synthesis and characterization of a model extracellular matrix that induces partial regeneration of adult mammalian skin". Proceedings of the National Academy of Sciences of the United States of America, 1989. 86(3): p. 933-7. 70. Zaleskas, J.M., et al., "Growth factor regulation of smooth muscle actin expression and contraction of human articular chondrocytes and meniscal cells in a collagen-GAG matrix". Experimental Cell Research, 2001. 270(1): p. 21-3 1. 47 48 APPENDIX A: A.1 FABRICATION OF COLLAGEN-GAG SCAFFOLDS Preparation of Collagen-GAG Slurry *Adapted from T. Freyman (Ph.D. Thesis, Massachusetts Institue of Technology, 2001) Summary A co-precipitate of type I collagen and chondroitin-6-sulfate is prepared by blending in the presence of acetic acid. The slurry is cooled during blending in order to keep the collagen from denaturing to gelatin. The triple-helical tertiary structure of the collagen is maintained. Eguipment Granco overhead blender Cooler Peristaltic Pump Granco Co., Kansas City, MO Brinkman cooler model CR-2T, Brinkman Co., Westbury, NY Manostat Cassette Pump, CAT# 75-500-0.00, New York, NY Materials Acetic Acid (HOAc) Type I Collagen Glacial Acetic Acid, Mallinckrodt Chemical Co., Paris, KY Chondroitin 6-sulfate Sigma # C-4384, from shark cartilage Dry microfibrillar bovine tendon collagen, Integra Lifesciences, Plainsboro, NJ Methods 1. Turn on cooling system and allow to cool to 4'C. 2. Prepare 0.05M acetic acid (HOAc) solution: 8.7 ml HOAc in 3 L dH20 This solution has a shelf ife of approximately 1 week. 3. Blend 3.6 g dry microfibrillar bovine tendon collagen with 600 ml of 0.05 M acetic acid on HIGH speed setting for 90 minutes at 4'C. 4. Prepare chondoitin 6-sulfate solution: 0.32 g chondroitin 6-sulfate in 120 ml acetic acid, stir for approx 30 minutes to mix well 5. Calibrate peristaltic pump to 40 ml/5 min. 6. Add 120 ml chondroitin 6-sulfate solution dropwise to the blending collagen dispersion over 15 minutes using the peristaltic pump (maintain blender at 4'C). 7. Blend 90 additional minutes on HIGH speed at 4'C. 8. Degas in series of vacuum flasks until bubbles are no longer present. 9. Store at 4'C. Slurry is good for approximately 4 months. If stored for more than one week, blend on LOW speed 15 minutes prior to using. 49 A.2 Freeze-Drying *Adapted from H. Breinan (Ph.D. Thesis, Massachusetts Institute of Technology, 1998) Summary The collagen-GAG slurry is frozen, causing formation of ice crystals. Reducing the pressure then causes the crystals to sublimate, resulting in a porous structure. The size of the crystals, and thus the size of the pores, is determined by the freezing temperature. Equipment VirTis Genesis Freeze-Drier (Virtis, Gardiner, NY) Methods 1. Turn on freeze-drier. 2. Turn on freeze switch. 3. Ensure that the plug is in the drain and turn on condenser. 4. Wait for the shelf temperature to reach -43'C (approx. I hour). 5. Pipette slurry into freeze-drier trays, being careful not to introduce air bubbles. 6. Place slurry into the freeze-drier and allow to freeze for 1.5 hours. 7. When slurry has frozen, turn on vacuum. Make sure that the door is sealed, chamber release is off, and that the condenser is less than -50'C. 8. Wait for the vacuum to reach less than 200 mtorr. Set the temperature to 00 C and turn on the heat switch. Allow to sublimate overnight. 9. Set temperature to 20'C and turn off the freeze switch. 10. When temperature reaches 20 (approx 30 minutes), turn off heat, vacuum, and condenser. 11. Turn on chamber release, remove sample, open the drain plug, and turn off power. A.3 1. Dehydrothermal (DHT) Cross-linking Prepare "envelopes" from aluminum foil large enough to hold matrix sheets. 2. Remove the collagen-GAG matrix sheets from the freeze-drier trays and place them in the aluminum foil envelopes. Be careful not to crush matrices. 3. Place open envelopes in the DHT oven. 4. Close oven. Turn VACUUM knob to ON and CHAMBER RELEASE knob to OFF. 5. Turn on vacuum pump. Make sure oven temperature is at 105'C and vacuum is <50 torr. Leave for 24 hours. 6. Turn of vacuum pump. Turn of the VACUUM knob and turn CHAMBER RELEASE to ON. 7. Once the pressure returns to atmospheric, open door and quickly seal envelopes. Store matrices in sealed packets in a dessicator. 50 APPENDIX B: B.1 CELL ISOLATION AND CULTURE Tissue Harvest OR Supplies Scalpel blades Scalpel handles Betadine Betadine sponges Sterile towels/wraps Saw Forceps Gloves Face masks Cooler with ice Method Shave hair from limb and scrub with betadine. Remove either entire limb or only the joint. If removing only the joint, first remove skin, then saw through bones on either side of the joint a distance far enough away from the joint to ensure that the joint capsule remains closed. If possible it is usually desirable to ligate large blood vessels prior to cutting. Wrap joint in sterile towels and place on ice in cooler. Lab Supplies Plastic basin Betadine Sterile scalpel blades Sterile scalpel handles Sterile Sterile Sterile Sterile cutting board Petri dishes forceps gloves Complete PBS Complete medium Tissue culture flasks Methods 1. Prepare cutting board and scalpels. Fill petri dishes with PBS. 2. Place joint in plastic basin and soak in betadine. 3. Put on sterile gloves. 4. Place joint on cutting board. 5. Open joint capsule by cutting through patellar tendon and making a cut next to the tendon into the joint space. 6. Remove synovium and place in petri dishes with PBS 7. Set aside samples of synovium for histology. 8. Dice remaining synovial tissue and transfer to 25 cm 2 culture flasks. 9. Place just enough medium in flasks to just cover the tissue, but cause it to float. 10. Pace flasks in incubator for 2-3 days to allow tissue to attach. 11. Place synovium samples allocated for histology in 10% formalin. 51 B.2 Passaging Cells Materials Complete medium Trypsin PBS (Phosphate-buffered saline) Glass pipettes Vacuum setup Sterile plastic pipettes Centrifuge tubes Tube holders Tissue culture flasks Methods 1. Warm the medium, trypsin, and PBS in 37'C water bath. 2. Remove medium from flasks with vacuum pipette (change pipettes for different animals). 3. Rinse with PBS (enough to cover bottom of flask, ~ 10 ml for 75 cm 2 flask). Trypsin will not detach the cells if it has come into contact with the medium. 4. Remove PBS and add trypsin (0.5 ml per well of 6 well plate, 2 ml for 25 cm 2 flask, 5 ml for 75 cm 2 flask). 5. Place in incubator for 5 minutes (unless otherwise instructed). 6. Remove from incubator and tap on the sides of the flask to loosen the cells. Check under microscope to ensure the cells are no longer attached. If they are, return them to the incubator and check each minute until they are unattached. 7. Once the cells are floating, return to the hood and add complete medium to inactivate the trypsin (1.5 ml per well of 6 well plate, 3 ml for 25 cm 2 flask, 10 ml for 75 cm 2 flask). 8. Using a sterile plastic pipette, transfer the medium/trypsin/cell suspension to a centrifuge tube. At this point you can combine the contents of the flasks if they are from the same sample. 9. Balance the tubes and centrifuge at 1500 rpm for 10 minutes. 10. Once you have the cell pellet at the bottom of the tube, draw off the medium with the vacuum pipette (be sure not to suck up the cells!!!). 11. Resuspend and count the cells (see Cell Counting protocol). While counting, centrifuge the cell suspension a second time to ensure all trypsin has been removed. 12. Decant medium from second centrifugation and resuspend at desired seeding density. Transfer to culture flasks and add complete medium to bring the flasks up to final volume. 52 B.3 Cell Counting *Adapted from Current Protocols in Cell Biology Materials Complete medium Trypan Blue Cell counting slide 70% ethanol Micropipetters Calculator Cell counter Pipette tips Pipette Aid coverslip Methods 1. Clean surface of hemacytometer and coverslip with 70% alcohol. 2. Wet edge of coverslip slightly with tap water and press over grooves on hemacytometer. The coverslip should rest evenly over the silver counting area. 3. Beginning with a cell pellet, suspend the cells in a known amount of complete medium 4. Collect a 100 pLd sample from the cell suspension and dilute with trypan blue (1:2 dilution if few cells are expected, 1:5 or 1:10 if a large number is expected). grid load cell suspension 1mm 5.Mix well, and collect 15 pl of suspension in a micropipette tip. 6. Load the cell suspension into the hemacytometer, allowing it to be drawn under the coverslip by capillary action. Load just enough cell suspension to reach the edges of the silvered surface. Do not overfill as this may change the volume and make the count inaccurate. I 1 7.Place hemacytometer on microscope stage, remove yellow glass filter, and view with standard lOx objective. 8. Count cells in each of the four corner squares and the central square (clear cells are viable, stained cells are dead). Count cells that lie on the top and left lines but not those on the bottom or right lines of each square in order to avoid counting the same cells twice for adjacent squares. Repeat counts for other counting chamber. A maximum cell count of 20 to 50 cells per lxl-mm square is recommended. When a count of living cells is complete, count the number of dead cells in order to report viability. 1mm 0 1 1 1 1 Picture from Current Protocols in Cell Biology Online 9.Calculate total cell number from the following: T= xD x104 Ns x V T = Total number of cells in suspension Nc = Number of cells counted. Ns = Number of 1mm squares counted. D = Dilution factor V = Volume of media used to suspend cell pellet. The number 104 is the volume correctionfactorfor the hemacytometer: each square is lx] mm and the depth is 0.1 mm. 10. Begin preparing samples for culturing and/or protein extraction 53 B.4 Freezing Cells Materials Complete Medium Dimethyl sulfoxide (DMSO) Sterile Filter Pipettes Sterile cryogenic tubes Methods 1. Determine amount of medium needed (1 ml per 2x10' cells) 2. Prepare solution of 10% DMSO in complete medium. Sterilize the medium +DMSOsolution by filtering through 0.22 or 0.45 pm sterilefilter, or by autoclavingDMSO prior to adding it to the medium. 3. Adjust cell concentration to 2x10 6 cells/ml of complete medium + DMSO solution. 4. Store in cryogenic tubes (3ml per 5 ml tube, 1 ml per 2 ml tube) in the -20'C freezer for 2-4 hours (longer time in this range is preferable), then transfer to -70'C for storage. B.5 Thawing Cells Methods 1. Place cells directly into a 37'C water bath. Agitate gingerly while cells thaw for 40-60 seconds. 2. When defrosted minimally (see liquid around outer edges) add a drop of complete medium. 3. Wait a minute and add another drop of medium. Repeat until tube is full. This insures that the cells thaw into the medium. 4. Transfer the cells to a 50 ml tube and wash them clean of medium+DMSO 2x for 10 minutes in the centrifuge. 5. Count the cells, and resuspend at the proper concentration. Cells should be cultured at least 3-4 days before being used for experimentation (or before changing medium, depending on when they attach). 54 B.6 Cell-Seeding of Collagen-GAG Scaffolds *Adapted from D. Hastreiter (Ph.D. Thesis, Massachusetts Institute of Technology, 2002) 1. Prepare and autoclave 2% (w/v) agarose (SeaPlaque Agarose, BioWhittaker #50101) 2. Pipette 1.5 ml agarose into each well of 12-well plates and allow to set for at least 4 hours at 4'C. Warm in incubator at 37'C prior to use. 3. Pre-wet matrices in PBS for 1 hour. 4. During this time, passage cells from flasks. Suspend cells at desired concentration. Use 50 pl for each 9 nm disc. For example, for a desired concentration of 2 x 106 cells/disc, the concentration should be 4 x 107 cells/ml. 5. Transfer matrices to medium for 10 minutes. 6. Briefly dry matrices on sterile filter paper (allow enough time for liquid to be drawn out, but do not allow matrices to dry completely) 7. Transfer matrices to warmed 12-well plates. 8. Pipette 25 pl onto one side of each matrix. After 10 min., flip matrices and pipette 25 pl onto the opposite sides. 9. Place matrices in incubator for 2 hours. 10. Add 0.5 ml medium (or enough to just cover matrices) to each well. 11. The following day, add another 1.0 ml medium to each well. B.7 1. Cell-Pellet Cultures Suspend 2 x 10 5 cells in medium in a 15 ml centrifuge tube. Centrifuge at 500g for 10 minutes. 2. Place tubes in incubator at 37'C overnight. Cells should form a free-floating pellet within approximately 16-20 hours. 3. Culture as desired, being careful not to break pellets when changing media. 55 56 APPENDIX C: C.1 BIOCHEMICAL ASSAYS Western Blot for detection of SMA C.1.1 ProteinExtraction Lysing Buffer 1% Sodium Dodecyl Sulfate (SDS) Sigma #L-4509 1 mM Sodium Orthovanadate (SO) 1 mM Phenylmethanesulfonyl fluoride (PMSF) 10 % Glycerol Sigma #S6508 Sigma #P7626 Sigma #G8773 To make 50 ml: 45 ml of 1% SDS 5 ml of 10% glycerol 100 91 of 100 mM stock SO 250 1d of 200 mM stock PMSF Methods *Begin with cell pellet after centrifugation 1. Rinse pellet with PBS after medium is drawn off. Spin for 10 min. and remove PBS 2. Suspend cells in lysing buffer at a concentration of at least 2x 106 cells/ml. 3. Transfer cell suspension to microcentrifuge tubes. 4. Shake tubes on shaker in cold room (4*C) for 10 minutes. 5. Centrifuge at 4*C for 20 minutes at 14,000 rpm. 6. Draw off supernatant and put into a new microcentrifuge tube. Discard pellet 7. Label and store at -20*C until evaluation. 57 C. 1.2 Protein Assay Summary Based on the Bradford method (Analytical Biochemistry, 1976. 72: p. 248-54), this procedure involves the additions of an acidic dye to a protein solution. The binding of the dye to the protein results in a differential color change of the dye, which is measured at 595 nm with a spectrophotometer. Equipment Spectrophotometer LKB Biochrom Ultraspec 4050 Materials Bovine Serum Albumin (BSA) BioRad #500-0007 Protein Assay Dye Cuvettes BioRad #500-0006 Fisher #14-386-21 Methods 1. Dilute the BSA stock solution (1.5 pg/pl) to a 1:10 with dH 20 for a final concentration of 0. 15 ptg/ptl. Prepare25 pl BSA with 225 l dH20for a total of 250 ,l. 2. Prepare gradient for standard curve. Each sample should be a total of 2 ml. The BSA standard is linear within the range of 0.2-0.9 mg/ml. d BSA (0.15p4g/pl) 0 (blank) 10 (1.5 pg) 20 (3.0 4g) 40 (6.0 4g) 60 (9.0 Vtg) 80 (12 pg) pl dH 20 1600 1590 1580 1560 1540 1520 p1 Dye (BioRad) 400 400 400 400 400 400 3. To prepare samples, add 20 pl of supernatant to 1580 pl of dH 20. Then add 400 ml of dye and mix. 4. Incubate at room temperature for 5-10 minutes. 5. Turn on spectrophotometer and allow to warm up. Add the standard set with the blank first (blue holder). 6. Read wavelength at 595 nm. To read the next sample press the "cell number" button. 7. Read samples. Leave the blank in and read it first, setting the reference to zero each time. 8. Generate a standard curve in Excel to determine how much protein is in each sample. 58 C.1.3 SDS-Polyacrylamidegel electrophoresis (PAGE) Summary One-dimensional gel electrophoresis separates proteins as they move through the pores of the gel, the size of which are determined by the concentration of acrylamide. Proteins are denatured by heat and detergent (SDS) to ensure separation on basis of size, eliminating effects of charge and shape. SDS also applies a negative charge to the proteins, which causes them to move through the gel toward the anode when the electric field is applied, the smaller proteins moving more rapidly through the porous matrix than larger proteins. After separation, the proteins are electrophoretically transferred ("blotted") onto a PVDF membrane for immunoprobing. (see Laemmli, 1970, Nature 227:680) Materials 2 mercaptoethanol Acrylamide Ammonium Persulfate BIS Bromophenol Blue Coomassie Blue Dry Milk Filter Paper (for blots) Glycerol Glycine SDS Primary Antibody PVDF Membranes Secondary Antibody TEMED TBS Trizma Base Tween 20 X-ray Film 20X Lumiglo Sigma M-7154 Sigma A-3553 Fisher A-682-500 Sigma B-8026 Sigma B-8522 BioRad 170-6404 BioRad 162-0118 Sigma G-8773 Sigma G-8898 Sigma L-4509 Sigma A-2547 BioRad 162-0176 Sigma A-2304 Sigma T-9281 Sigma T-6664 Sigma T-1503 Sigma F-5513 Cell Signal Tech. Cat# 7003 Prepared Solutions 30% Acrylamide-bisacrylamide Acrylamide BIS dH 20 29.2 g 0.8 g 100 ml 1.5% Ammonium Persulfate Ammonium Persulfate 150 mg dH 20 Note: Filter through #1 Whatman filter paper, store at 41C. Good for 1 month Note: Store at 4'C, good for 1 week 10 ml Resolving Buffer Tris base (3.0M) dH 20 46.8 g 100 ml Note: Filter through #1 Whatman filter paper, store at 7.9 g 100 ml Note: Filter through #1 Whatman filter paper, store at 4'C. pH = 8.8 Good for 1 month Stacking Buffer Tris base (0.5M) dH 20 Sample Buffer Tris base (0.5M, pH 7.0) 1.0 ml Glycerol 0.8 ml 10% SDS 1.6 ml 2ME 0.4 ml Dye 0.2 ml 4'C. pH = 6.8 Good for 1 month Notes: 1. Store at -20'C 2. 2ME = 2 mercaptoethanol, in hood in biochem lab (reduces di-sulfide bonds) 3. Dye = 1% bromophenol blue 59 Running Buffer (10X stock solution) Tris base 30.3 g 144.0 g Glycine SDS dH 20 Note: pH = 8.3, Store at room temperature 10.0 g 1.0 L Transfer Buffer (5X stock solution)*** Notes: Tris 15.15 g 1. Store at room temperature Glycine 72.25 g 2. ***For dilution to Ix, add 20% methanol.*** dH 20 800 ml (160 ml 5X buff, 480 ml dH 2 0, 160 ml methanol) TBS-T Tween 20 1.0 ml TBS 1.0 L Methods - SDS PAGE 1. Assemble the gel-casting apparatus. Fill ice tray for blot transfer and place in freezer. 2. Prepare the resolving gel. Load 7 ml of gel solution in each cassette. Overlay with a small amount of dH 20. Amounts will vary depending on the %Tofthe gelyou desire. The following is for a 12% gel. Amounts are enough for 2 gels - dispose of extra in trash, not the sink. Resolving gel dH 20 Acryl-bis Resolving buffer 8.2 ml 8.0 ml 2.5 ml 10% SDS 200 ptl AP 1.0 ml TEMED 20 jil Add TEMED underfume hood. 3. Begin boiling water for samples. 4. Prepare samples, control, and marker. For each sample, combine 24 pl of sample buffer with 5 gg of protein, then bring to a total of 48 pl with dH20. Add sample buffer underfume hood 5. When the resolving gel is set (approx. 20-30 min.), remove layer of dH 2O using filter paper. 6. Prepare the stacking gel. Load stacking gel solution and insert combs. Stacking Gel: dH 20 Acryl-bis 5.6 ml 1.25 ml Stacking buffer 10% SDS AP 2.5 ml 100 pl 500 pl TEMED 10 jd Add TEMED underfume hood 7. Heat the samples in boiling water for 5 minutes, then centrifuge in cold room for 10 minutes at the highest speed (14,000rmp). 8. Once the gels are solid, remove the combs and move the cassettes to the gel-running chamber. 60 9. Fill the inside of the chamber with lx Running buffer. Fill the outside of the chamber with the buffer to at least the first set of knobs. 10. Use the special loading pipette tips to load the samples in the gels. Before loading samples, fill each well with running buffer to remove any bubbles. 11. Run the gel for approx. 2 hr. at 90V. It is finished when the sample buffer has reached the bottom of the gel, so check periodically. 12. Remove the gel by using one of the spacers to carefully pry the glass plates apart. Cut away the stacking portion of the gel. Mark the gel in a way that such that the orientation will be preserved. 61 C. 1.4 Blot Transfer Methods 1. Cut PVDF membranes and filter paper to same dimensions as gel. 2. Briefly wet membranes in pure methanol. Be sure not to touch membranes with bare hands as oilfrom the hands can block transfer. 3. Soak the gel, filter paper, and sponges in lx Transfer buffer (*** add methanol, see I' page ***) for 10 minutes to equilibrate the gel. Be sure to keep track of which gel is #1 and #2. Gel equilibrationis requiredto prevent a change in the size of the gel duringtransfer. 4. Assemble the transfer sandwich according to the following diagram. Keep the cassette submerged in transfer buffer to avoid air bubbles and prevent the membrane from drying. Gently rub out any bubbles between the gel and the membrane using a stir rod. Clear/White Plate Sponge Filter Paper Membrane Gel Filter Paper Sponge Black Plate 5. Load the cassette into the transfer chamber. Black faces black. Load ice tray into chamber. 6. Add the remaining lx TBS for a total of 1 L. 7. Run for 1 hour at 100 V. If desired, do this step in the cold room at 4' C. 8. Remove membranes and wash in lx Transfer buffer for 5 minutes. 9. Stain gels with Coomassie Blue to verify transfer of proteins to the membrane. Membranes can be driedand stored in resealableplastic bags at 4 Cfor 1 year or longer at this point. Priorto furtherprocessing,dried PVDF membranes must be placed into a small amount of 100% methanol to wet the membrane, then in distilled water to remove the methanol. ( Current Protocols in Cell Biology) If proceeding to immunoblotting and detection the following day, place membranes in blocker (5% dry milk) overnight at 4'C. To prepare blocker: 5g dry milk to 100 ml TBS. Mix well. 62 C.1.5 Immunoprobing and Visualization of Blotted Proteins Summary Immobilized proteins are probed with antibodies to the desired antigen under investigation. All open protein-binding sites on the membrane are first filled by immersion in blocking buffer containing nonreactive protein (dry milk). Membranes are then incubated with a primary antibody against the desired antigen, followed by an enzyme-antibody conjugate against the primary antibody (peroxidase). A chemiluminescent reagent that reacts with the conjugated enzyme on the secondary antibody is applied, and X-ray film exposed by the luminous bands. Methods - Probing 1. If membranes have been stored, briefly wet membranes in pure methanol, then wash with distilled water. 2. Place membranes in blocker (5% dry milk) for 30 minutes on rocker. To prepare blocker: 5g dry milk to 100 ml TBS. Mix well. 3. Incubate with primary antibody for 2 hours at room temperature on rocker. 1:400 dilution. Add 50 d of primary antibody to 20 ml Ix TBS-T. The antibodies may stored at 4 C and reused, but do not store more than 24 hours. 4. Rinse in Ix TBS-T for 10 minutes, 3 times. 5. Incubate with secondary antibody for 1 hour at room temperature on rocker. 1:5,000 dilution. Add 4 pl secondary antibody to 20 ml Ix TBS-T. 6. Rinse in Ix TBS-T for 10 minutes, 3 times. Methods - Detection 1. Prepare LumiGlo solutions A and B. You need a 1 ml of each solution for each membrane. Do not mix A and B together until you are ready to use them. Once washing is complete, thoroughly mix A and B together and put 2 ml on each membrane. Solution A: Mix 100 ptl A to 2 ml dH 2 0. Solution B: Mix 100 d B to 2 ml dH20. 2. Incubate for 2 minutes at room temperature. 3. Wrap membranes in saran wrap, carefully smooth out any air bubbles 4. Expose film in dark room (Thorn 1224C) and develop. Adjust exposure time as needed. 63 C.1.6 DensitometricAnalysis *This protocol is adapted from Scion Image Manual 1. Open Scion Image software and load scanned image file 2. Click Load Macros in the Special menu. Load "GelPlot2" from the Macros directory. 3. Use the rectangular selection tool to outline the lanes 4. Select Mark First lane in the Special menu. 5. Select Plot Lanes in the Special menu. 6. Use the line drawing tool to draw base lines and drop lines so that each peak defines a closed area. 7. Measure the areas of the peaks by clicking inside each one in succession with the wand tool. 8. Use the text tool while holding "Scroll Lock" to label peaks, in reverse order, with the area measurements. The area measurements are also recorded in tabular form that can be exported to a spreadsheet. 64 C.2 DNA Analysis C.2.1 PapainDigestion *Samples should be lyophylized and their mass determined prior to digestion* Materials Sodium Phosphate, monobasic Sodium Phosphate, dibasic Fisher #S369 Fisher #S373 Fisher #S311 Sigma #C1276 Sigma #P3125 Disodium EDTA L-Cystein HCL Papain Solutions 0.5 M Monobasic stock NaH 2PO 4 *H 20 dH 20 6.9 g 0.5 M Dibasic stock Na2HPO 4 *7H 20 dH 20 13.4 g 100 ml 100 ml Papain buffer Dibasic stock Monobasic stock L-Cysteine HCL 2.46 ml 17.54 ml 87.82 mg Disodium EDTA 186.12 mg Papain 0.5 ml (stock is 25 mg/ml) dH 20 80 ml Methods Place lyophylized matrices in microcentrifuge tubes and add 1 ml papain buffer per tube. Place tubes in 65'C waterbath overnight. C.2.2 FluorometricQuantificationofDNA Equipment TKO 100 Fluorometer, Hoeffer Scientific Instruments, San Francisco, CA Materials Tris Base Na 2EDTA NaCl Hoecsht 33258 Calf thymus DNA Fisher Fisher #S311 Fisher #S271 Sigma #B2883 Sigma #D-3664 Solutions lOX TNE buffer Tris Base Na2EDTA 100 mM (12.1 g/L) 10 mM (3.7 g/L) NaCl 1.0 M (58.4 g/L) pH to 7.4, sterile filter, store at 4'C Hoechst dye stock solution Hoechst 33258 10 mg Note: carcinogenic and light sensitive 65 dH 20 (sterile) 10 ml Working dye solution 1OX TNE dH 20 10 ml 90 ml -+Filter through 0.45 pm filter -+Filter through 0.45 pm filter Hoechst stock solution 10 pl Methods 1. Turn on fluorometer and allow it to warm up for 15 minutes. Make sure the "scale" knob is adjusted to 50% sensitivity (approx. 5 clockwise turns from fully counter-clockwise position). 2. Prepare DNA standard in duplicate (or triplicate). ig of DNA pl of DNA 10 5 2.5 1 jU from 1 mg/mi stock 50 pil from previous 50 pl from previous 99 50 50 1.25 50 pd from previous 50 0.625 0.3125 50 l from previous 50 pl from previous 50 50 0 0 50 4l of PBE 3. Add 20 p1 of each standard to a cuvette containing 2 ml of working dye solution. 4. Once the machine is zeroed, add the 10 pg standard and adjust the scale knob to read 100. Do not readjust the scale knob once the standard curve has been established. 5. Read samples and make certain it is linear (with R2<0.95). 6. Run samples in duplicate. Add 50 p1 of sample to 2 ml of working dye solution. Adjust sample amount as necessary if readings are too high or low. 66 APPENDIX D: D.1 HISTOLOGY AND IMMUNOHISTOCHEMISTRY Paraffin Embedding *Adapted from H. Breinan (Ph.D. Thesis, Massachusetts Institute of Technology, 1998) Epuipment Tissue Tek VIP 1000 model 4617 Tissue Tek tissue embedding center Materials Clearing Solutions: Xylene (Fisher #X5, or substitute, such as Histosolve or Citrisolve) Paraffin (Fisher # 23-021-400) Methods 1. Dehydration and infiltration A. Tissue specimens are dehydrated and infiltrated by machine (Tissue Tek program 4), with solutions changed automatically as follows: 50% ethanol 70% ethanol 80% ethanol 95% ethanol 100% ethanol Clearing solution Paraffin Paraffin 1 hour 1 hour 1 hour 2x 1 hour 3x 1 hour 2x 1 hour 1 hour 2x 30 min. Room Room Room Room Room Room temperature temperature temperature temperature temperature temperature 59 0 C 59 0 C B. Fragile collagen matrices and cell pellet cultures are dehydrated by hand. For pellet culture specimens, the following solutions are pipetted in and out of 24 well plates. Matrices specimens are placed in plastic tissue cassettes, which are then placed in the following solutions: dH 20 50% ethanol 70% ethanol 80% ethanol 95% ethanol 100% ethanol Clearing solution Paraffin 2. 3x 30 min. 30 min. 30 min. 30 min. 2x 30 min. 3x 30 min. 1 hour 2x 1 hour Room Room Room Room Room Room Room temperature temperature temperature temperature temperature temperature temperature 59 0 C Embedding. " * * * " Remove specimens from tissue cassettes Partially fill stainless steel or plastic mold with molten paraffin Place specimen in mold with desired orientation and transfer to cooling plate briefly Place tissue cassette on the mold and affix with additional paraffin Place in freezer overnight prior to removing from mold 67 Hematoxylin and Eosin (H & E) Staining D.2 Summary Formalin fixed, paraffin embedded sections are stained with H&E for visualization of structure. Hematoxylin stains acidic portions deep blue (such as cell nuclei rich in DNA and RNA). Eosin stains basic substances pink (the collagenous ECM). Solutions Hematoxylin Harris Hematoxylin Solution, Sigma Cat# HHS-128. Filter 200ml of stock solution into staining dish. Acid Alcohol 200ml of 70% ethanol (in dH20) + 0.5 ml HC1 Ammonia water 200 ml dH20 + 5-10 drops ammonium hydroxide, pH around 10.0 Eosin Eosin Y Solution Aqueous, Sigma Cat# HTI 10-2-128 Other Materials Cytoseal 60 Electron Microscopy Sciences Cat# 18006. Methods Paraffin Sections 1. Deparaffinize and Rehydrate Xylene (or substitute) 100% ethanol 100% ethanol 95% ethanol 80% ethanol 70% ethanol dH 20 2 x 5 minutes 10-20 dips 10-20 dips 10-20 dips 10-20 dips 10-20 dips 10-20 dips 2. Harris hematoxylin - 10 minutes. 3. Rinse in tap water, approximately 1 min. running or swishing until almost clear. 4. Acid alcohol. Dip quickly 5-10 times, 20-30 sec. total. 5. Rinse in tap water until foaming stops, approximately 30 sec. 6. Ammonia water. Quick dips (5 or so) until blue. 7. Rinse in tap water approximately 1 min. 8. Eosin, 45-60 sec. 9. Rinse in tap water, 1-2 min. 10. Dehydrate 70% ethanol 80% ethanol 95% ethanol 100% ethanol 100% ethanol Xylene (or substitute) 10-20 dips 10-20 dips 10-20 dips 10-20 dips 10-20 dips 2 x 5 minutes 11. Air dry. Coverslip with Cytoseal 68 D.3 Immunohistochemical Staining of a-Smooth Muscle Actin Summary Deparaffinized and rehydrated sections are incubated with trypsin to uncover antigenic sites, followed by quenching of endogenous peroxidase with H 2 0 2 . After blocking of nonspecific binding, primary antibody against the desired antigen (SMA) is applied. A biotinylated secondary antibody binds to the primary antibody, and is labeled with peroxidase via avidin- biotin binding. The bound peroxidase is then reacted with an AEC chromagen. Solutions **Note: Amounts will vary depending on number of slides being stained** Phosphate Buffered Saline (PBS) (Sigma # P-3813) 1 package to 1 L dH 20, make 2L for 14 slides 0.1% Trypsin (Sigma # T-7409) 0.01g trypsin 10 ml PBS Store at 4'C until use, dessicate 3% Hydrogen Peroxide (H 2 0 2 ) (Sigma # H-1009) I ml 30% H 2 0 2 9 ml d1120 Stored at 4'C 30% Goat Serum (Sigma # G-9023) Stored at -20 0 C 0.3 ml Goat Serum 0.7 ml PBS Primary Antibody - Mouse Monoclonal Anti-c-Smooth Muscle Actin (Sigma #A2547) 5 pl primary antibody 2 ml PBS Stored at -20'C Use a 1:400 dilution Secondary Antibody - Biotinylated Goat Anti-Mouse Immunoglobin (Sigma # B715 1) 10 pl secondary antibody Stored at -20'C 2 ml PBS Negative Control -Mouse Serum (Sigma # M-5905) 5 pl mouse serum 4 ml PBS Extravadin-Conjugated Peroxidase (Sigma # E2886) 20 gl peroxidase I ml Substrate 1 1 1 1 Use a 1:200 dilution Stored at -20'C Dilute to same protein concentration as diluted primary antibody Stored at 4'C Use a 1:50 dilution PBS Reagent (Zymed # 00-2007) ml dH 20 drop substrate buffer (bottle A) drop chromogen solution (bottle B) drop hydrogen peroxide (bottle C) Stored at 40 C Keep away from light Other Materials Mayer's Hematoxylin Solution (Sigma # MHS-16) Glycerol Gelatin (Sigma# 49927) 69 Methods ***DO NOT TOUCH THE SPECIMENS NOR LET THEM DRY OUT AT ANY POINT DURING THIS PROCESS*** 1. Deparaffinize and rehydrate via the following baths Xylene (or substitute) 100% EtOH 100% EtOH 95% EtOH 80% EtOH 70% EtOH PBS PBS 2. 1 2 2 2 2 2 2 2 hour or overnight, stir gently minutes minutes minutes minutes minutes minutes minutes Wipe off PBS from non-sample areas with Kim Wipe. DO NOT TOUCH SAMPLES. Circumscribe the samples with a PAP pen. 3. Drop trypsin solution onto samples using disposable pipette. Incubate 1 hour at room temperature. 4. During this time, prepare 3% H 2 0 2 , 30% goat serum, primary antibody, and mouse serum. 5. Wash 2x in PBS, 2 minutes each. 6. Incubate with 3% H 2 0 2 for 5-10 minutes. 7. Wash 2x in PBS, 2 minutes each. 8. Incubate in 30% goat serum for 10 minutes. 9. Wipe off serum, but do not wash in PBS. 10. Apply primary antibody or negative control and incubate for 2 hours. 11. Rinse individual sections separately with PBS using a pipette to prevent contamination of the negative control with the primary antibody. 12. Wash 2x in PBS, 2 minutes each. 13. Incubate with secondary antibody for 20 minutes. 14. Wash 2x in PBS, 2 minutes each. 15. Incubate with extravidin peroxidase for 20 minutes. During this time, prepare substrate reagent and warm the glycerol gelatin. 16. Wash slides in PBS for 2 minutes, then in dH 20 for 3 minutes. 17. Dry 5 slides quickly, then incubate with AEC substrate reagent. Check slides under microscope after 3 minutes. If darker staining is desired, leave in AEC for 1-2 additional minutes. 18. Wash slides in dH20 for 3 minutes. 19. Place slides in Mayer's hematoxylin solution for 20 minutes. 20. Rinse with running water for 20 minutes. 21. Coverslip with warmed glycerol gelatin. If gelatin hardens too quickly, place slides on 40 degree surface or in 57 degree oven for a few minutes to re-melt the gelatin. 70 D.4 Immunohistochemical Staining of Type II Collagen Solutions * *Note: Amounts will vary depending on number of slides being stained** Tris-Buffered Saline (TBS) (Sigma # T-6664) 1 package to 1 L dH 2 0. Protease XIV (a.k.a. Pronase E) (Sigma # P-5147) 0.01g Protease 10 ml Store at -20*C until use, dessicate TBS 3% Hydrogen Peroxide (H 2 0 2 ) (Sigma # H-1009) 1 ml 30% H 20 2 dH 20 9 ml Stored at 4*C 5% Horse Serum (Sigma # H-0146) 0.5 ml Horse Serum Stored at -20*C 10 ml TBS Primary Antibody - cIIcI mouse anti-chick type II Collagen monoclonal Antibody (Developmental Studies Hybridoma Bank, Iowa City, IA) 250 pl primary antibody 5 ml TBS Stored at -20'C Use a 1:20 dilution Secondary Antibody - Biotinylated Horse Anti-Mouse Immunoglobin (Vector # BK2000) 10 pA secondary antibody Stored at -20'C Use a 1:200 dilution 2 ml TBS Negative Control -Mouse IgG 2 A (Zymed #02-6200) Dilute IgG in TBS to same protein concentrationas in diluted primary antibody Stored at -20'C Avidin-Biotin Complex (ABC) staining kit (Vector #PK 4000) Stored at 4'C Diaminobenzidine (DAB) kit (Vector # SK-4100) Stored at 4'C Caution: Known carcinogen, handle carefully and dispose properly Other Materials Harris Hematoxylin Solution (Sigma # HHS-128) Permount (Fisher #SP15) 71 Methods ***Do not let the slides dry out during this process*** 1. Deparaffinize and rehydrate via the following baths Xylene (or substitute) 100% EtOH 95% EtOH 80% EtOH 70% EtOH TBS 2. 2 2 2 2 2 2 x 5 minutes x 2 minutes minutes minutes minutes x 2 minutes Wipe off TBS from non-sample areas with Kim Wipe. DO NOT TOUCH SAMPLES. Circumscribe the samples with a PAP pen. 3. Drop Protease solution onto samples using a disposable pipette. Incubate 1 hour at room temperature. During this time, prepare 3% H 2 0 2 , 30% horse serum, primary antibody, and negative control. 4. Wash 2x in TBS, 2 minutes each. Wipe slides afterward 5. Block with 5% horse serum for 30 minutes. 6. Remove serum, but do NOT wash afterwards. 7. Apply primary antibody or negative control and incubate for 1 hour. 8. Rinse individual sections separately with TBS using a pipette to prevent contamination of the negative control with the primary antibody. 9. Wipe slides, wash 2x in TBS, 2 minutes each. Wipe slides again 10. Incubate with secondary antibody for 45 minutes. During last 15 minutes, prepare ABC reagent and allow to stand for 30 min. 11. Wash 2x in TBS, 2 minutes each. Wipe slides afterwards. 12. Quench endogenous peroxidase by incubation in 3% H2 0 2 for 10 minutes. 13. Wash 2x in TBS, 2 minutes each. Wipe slides afterwards. 14. Incubate with avidin-biotin complex (ABC kit) reagent for 30 minutes. 15. Wash 2x in TBS, 2 minutes each. Wipe slides afterwards. 16. Dip slides in water. 17. Stain with DAB staining kit for 8-10 minutes. 18. Rinse in dH 20 for 3-5 minutes. 19. Counter-stain with Harris' hematoxylin for 10 minutes. 20. Rinse in running tap water. 21. Dip in acid alcohol 22. Rinse in running tap water. 23. Dip in ammonia water. 24. Rinse in water 25. Dehydrate (70%, 80%, 95%, 100%, 100%, xylene, 2 minutes each). 26. Air dry and coverslip with Permount. 72