Chapter 25 Gas-Liquid and High-Performance Liquid Chromatography

Chapter 31
Gas Chromatography
GAS-LIQUID CHROMATOGRAPHY
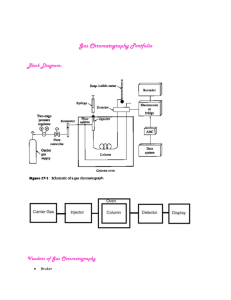
In gas chromatography, the components of a vaporized sample are fractionated as a consequence of being partitioned between a mobile gaseous phase and a liquid stationary phase held in a column. In performing a gas chromatographic separation, the sample is vaporized and injected onto the head of a chromatographic column.
Elution is brought about by the flow of an inert gaseous mobile phase. The mobile phase does not interact with molecules of the sample; its only function is to transport the sample species through the column.
Carrier Gas System
The gaseous mobile phase in gas chromatography must be chemically inert.
Helium is the most common mobile phase, although argon, nitrogen, and hydrogen are also used. These gases are available in pressurized tanks. Pressure regulators, gauges, and flow meters are required to control the flow rate of the gas. Pressures at the column inlet usually range from 10 to 50 psi(lb/in 2 ) and provide flow rates of 25 to 50 mL/min.
Sample Injection System
Column efficiency requires that the sample be of a suitable size and be introduced as a plug of vapor; slow injection or oversized samples cause band spreading and poor resolution.
Calibrated microsyringes are used to inject liquid samples through a rubber or silicone septum into a head sample port located at the head of the column. The sample port is ordinarily about 50 o C above the boiling point of the least volatile component of the sample.
Detectors
Detector devices for gas chromatography must respond rapidly to minute concentration. The solute concentration in the carrier gas at any instant is no more than a few parts per thousand. Moreover, the time during which a peak passes the detector is very short, which requires that the device be capable of exhibiting its full response during this brief period.
Desirable properties for a detector include high sensitivity, linear response, stability, reproducibility, wide temp.
range, high reliability, and uniform response for a wide variety of chemical species or, a predictable and selective response toward one or more classes of solutes and nondestructive of sample.
Flame-Ionization Detectors
The flame-ionization detector (FID) is the most widely used and generally applicable of all detectors for gas chromatography. Most organic compounds, when pyrolyzed in a hot flame, produce ionic intermediates that conduct electricity through the flame. Detection involves monitoring the current produced by collecting electrons and ions produced by the combustion process at biased electrodes. The FID exhibits a high sensitivity, a large linear response, and low noise. A limitation of the FID is that it destroys the sample during the combustion step.
Thermal Conductivity Detectors
The thermal conductivity detector, which was one of the earliest detectors for gas chromatography, still finds wide application. This device consists of an electrically heated source whose temperature at constant electric power depends on the thermal conductivity of the surrounding gas. The heated element may be a fine platinum, gold, or tungsten wire or, a small thermistor.
The advantages of the thermal conductivity detector are its simplicity, its large linear dynamic range, its general response to both organic and inorganic species, and its nondestructive character, which permits collection of solutes after detection. The chief limitation of the thermal conductivity detector is its relatively low sensitivity.
Electron-Capture Detectors
The electron-capture detector (ECD) has become one of the most widely use detectors for environmental samples because this detector selectively responds to halogen-containing organic compounds, such as pesticides and polychlorinated biphenyls. In this detector, the sample eluate from a column is passed over a radioactive
emitter, usually nickel-63. An electron from the emitter causes ionization of the carrier gas (often nitrogen) and the production of a burst of electrons.
Electron-capture detectors are highly sensitive and have the advantage of not altering the sample significantly.
Gas Chromatographic Columns and
Stationary Phases
Two types of columns are encountered in gasliquid chromatography, capillary , and packed .
The latter can accommodate larger samples and are generally more convenient to use than the former. Capillary columns have become of considerable importance because of their unparalleled resolution. In old days the vast majority of gas chromatography has been carried out on packed columns but capillary column is becoming popular day by day.
Column Thermostating
Reproducible retention time require control of the column temperature to within a few tenths of a degree. For this reason, the coiled column is ordinarily housed in a thermostated oven. The optimum temperature depends on the boiling point of the sample components. A temperature that is roughly equal to or slightly above the average boiling point of a sample results in a reasonable elution period. For samples with a broad boiling range, it may be necessary to employ temperature programming.
Liquid Phases for GC
Desirable properties for the immobilized liquid phase in a gas-liquid chromatographic column include (1) low volatility (2) thermal stability (3) chemical inertness and (4) solvent characteristics such that K and
values for the solutes to be resolved fall within a suitable range.
The retention time for an analyte on a column depends on its distribution constant, which in turn is related to the chemical nature of the liquid stationary phase. Clearly, to be useful in gasliquid chromatography, the immobilized liquid must generate different partition ratios for different sample components.
Application of Gas-Liquid Chromatography
Gas-liquid chromatography is applicable to species that are appreciably volatile and thermally stable at temperatures up to a few hundred degrees Celsius. An enormous number of compounds of interest to humans possess these qualities.
Consequently, gas chromatography has been widely applied to the separation and determination of the components in a variety of sample types.
Qualitative Analysis
Gas chromatography is widely used for recognizing the presence or absence of components in mixtures that contain a limited number of species whose identities are known.
The application to the qualitative analysis of complex samples of unknown compositions is limited.
This limitation has been largely overcome by linking chromatographic columns directly with ultraviolet, infrared, and mass spectrometers.
The resulting hyphenated instruments are powerful tools for identifying the components of complex mixture.
Quantitative Analysis
Gas chromatography owes its enormous growth in part to its speed, simplicity, relatively low cost, and wide applicability to separations. Quantitative
GC is based on comparison of either the height or the area of an analyte peak with that of one or more standards.
If condition are properly controlled, both of these parameters vary linearly with concentration. Peak area is independent of the broadening effects. Therefore, area is a more satisfactory analytical parameter than peak height.
Most modern chromatographic instruments are equipped with computers that provide measurements of relative peak areas.
Calibration with Standards
The most straightforward method for quantitative gas-chromatographic analyses involves the preparation of a series of standard solutions that approximate the composition of the unknown. Chromatograms for the standards are then obtained, and peak heights or areas are plotted as a function of concentration. A plot of the data should yield a straight line passing through the origin; quantitative analyses are based on this plot. Frequent standardization is necessary for highest accuracy.
The Internal-Standard Method
The highest precision for quantitative GC is obtained using internal standards because the uncertainties introduced by sample injection, flow rate, and variation in column conditions are minimized.
In this procedure, a carefully measured quantity of an internal standard is introduced into each standard and sample, and the ratio of analyte peak area (or height) to internalstandard peak area (or height) is used as the analytical parameter. For this method to be successful, it is necessary that the internalstandard peak be well separated from the peaks of all other components in the sample