THE CYTOSKELETON AND RELATED
STRUCTURES
The cell’s internal skeleton helps organize its
structure and activities
– A network of protein fibers make up the
cytoskeleton.
Tubulin subunit
Actin subunit
Fibrous subunits
7 nm
Microfilament
25 nm
10 nm
Intermediate filament
Microtubule
– Microfilaments of actin
• Enable cells to change shape and move
– Intermediate filaments
• Reinforce the cell and anchor certain organelles
– Microtubules give the cell rigidity
• And provide anchors for organelles and act as tracks for
organelle movement
– A typical plant cell has some structures that an
animal cell lacks
• Such as chloroplasts and a rigid cell wall
Nucleus
Rough
endoplasmic
reticulum
Ribosomes
Golgi
apparatus
Not in
animal
cells
Central
vacuole
Chloroplast
Cell wall
Mitochondrion
Peroxisome
Plasma membrane
Figure 4.4B
Smooth
endoplasmic
reticulum
Microtubule
Intermediate
filament
Microfilament
Cytoskeleton
Plant cells
• Are supported by rigid cell walls made largely of cellulose
• Connect by plasmodesmata, which are connecting channels
Walls of two adjacent
plant
cells
Vacuole
Plasmodesmata
Layers of one plant
cell wall
Cytoplasm
Plasma membrane
Figure 4.18A
– Tight junctions can bind cells together into leakproof
sheets
– Anchoring junctions link animal cells into strong
tissues
– Gap junctions allow substances to flow from cell to
cell
Tight junctions
Anchoring junction
Gap junctions
Extracellular matrix
Space between cells
Figure 4.18B
Plasma membranes of adjacent cells
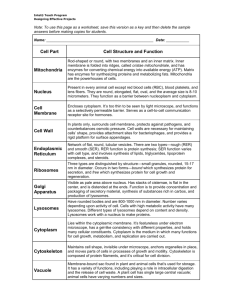
Human Organelle
Diseases/Problems
Cystic Fibrosis and the Cell Membrane
•Cystic fibrosis (CF) is caused
by a salt imbalance, making
mucus in the lungs and
digestive system extremely
thick.
•Caused by recessive gene;
about 20% of us are carriers
•Several new treatments,
including a healthy gene
introduced into the lungs in a
nasal spray, target the illness
at the cellular source.
Source:
http://learn.genetics.utah.edu/content/disorders/singlegene/cf/
Adrenoleukodystrophy (ALD) and Peroxisomes
• Cause: peroxisomes lacked the second most abundant
protein in the outer membrane of this organelle.
• Normally, the missing protein (a chaperone protein)
transports an enzyme into the peroxisome.
• Without the enzyme, fatty acids builds up in cells in the
brain and spinal cord, eventually myelin is depleted
(vital for nerve transmission). Death comes in a few
years.
• For many sufferers of ALD, eating a type of triglyceride
from rapeseed (canola) oil slows buildup of the very
long chain fatty acids for a few years, stalling
symptoms. But the treatment eventually impairs blood
clotting and other vital functions, and fails to halt the
progression of the illness.
Tay-Sachs Disease and Lysosomes
• More common in some ethnic communities, a
mutation of an enzyme in lysosomes
• In eyes, a telltale cherry red spot indicates the
illness
• the lysosomes, tiny enzyme-filled sacs, swell to
huge proportions.
• These lysosomes lack one of the forty types of
lysosomal enzymes, results in built up fatty
material on nerve cells.
• Sadly and commonly, the nervous system
continues to fail, and paralysis , then death
before the age of four.
Cystic Fibrosis & the Rough ER
• a prominent example of a disease caused by misfolded
proteins.
• CF is an ultimately fatal inherited disorder in which the
lack of a specific type of plasma membrane chloride
channel, the cystic fibrosis transmembrane regulator
(CFTR), causes the accumulation of a thick mucus that
compromises several organs, most notably the lungs
and pancreas.
• The misfolded CFTR protein becomes trapped within
the ER and is subsequently degraded.
•
Source:
http://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=2&ved=0CDMQFjAB&url=http%3A%
2F%2Fteachers.sduhsd.k12.ca.us%2Fmrall%2Fap%2520bio%2Fap%2520bio%2520homework%2520and%2
520ppts%2Fextra%2520credit%2FOrganellesdiseases.pdf&ei=3wJMU9nOC4MyAGp6YDAAw&usg=AFQjCNFQedV1c_P4wPuLdfuFjhN1RfVwQQ&sig2=ul8j-ICvcDSngsQO09rexA
ER stress
• induced by a variety of conditions such as
proteinaggregation, Ca2 depletion, glucose
deprivation, or fatty acid
• overload, can result in severe cell dysfunction
or death.
• It is an important feature of such neurodegenerative conditions as Alzheimer’s,
Huntington’s, and Parkinson’s diseases, as well
as heart disease and diabetes.
GOLGI APPARATUS
• The most commonly recognized Golgi-linked
diseases are a group of 15 congenital
disorders of glycosylation (CDG).
• Caused by mutations in genes that encode
glycosylation enzymes or glycosylation-linked
transport proteins
• a CDG is usually lethal by the age of 2.
• Symptoms include mental retardation,
seizures, and liver disease.
Nuclear Membrane problems
• defects in the nuclear envelope occur in the
genes that code for lamin, a cytoskeletal
component of the nuclear lamina, and emerin,
an inner membrane protein.
1. Progeria
• a fatal childhood
disease characterized
by premature aging of
the musculoskeletal
and cardiovascular
systems
• Has been linked to a
specific mutation in
the lamin A gene.
2. Emery-Dreifuss muscular
dystrophy
• caused by the absence or
mutation of the gene that
codes for emerin
• Symptoms include:
– a fragile nuclear membrane
– altered regulation of DNA
replication and transcription
– and low tolerance to
mechanical stress.
Lysosomes & Peroxisomes
• lysosomal storage diseases (LSD)
• caused by the absence of one or more lysosomal
enzymes
• Examples:
– Tay-Sachs and Gaucher’s, as well as Pompe’s disease
(glycogen storage disease type II), are caused by the
absence of a single enzyme. Death occurs in early
childhood.
– In I-cell disease, the import of all lysosomal enzymes into
lysosomes in certain organs is defective. In affected cells,
the enzymes are instead secreted into the extracellular
matrix. Symptoms include mental deterioration, heart
disease, and respiratory failure.
Mitochondrial problems
• High rates of mutation, since it
is an ancient bacterium
• Mutation in proteins that guide
mitochondrial division create
dissimilar daughter
mitochondria
• This increases risk of inheriting
mutations that are harmful to
mitochondrial function.
• Examples: exercise intolerance,
chronic fatigue
• Examples: diabetes, Parkinson’s,
Alzheimer’s
© 2005 Nature Publishing Group Taylor, R. W. et al. Mitochondrial DNA mutations in
human disease. Nature Reviews Genetics 6, 394 (2005). All rights reserved.
Mitochondria & Cancer?
• In 1998, a link between colorectal cancer and
somatic mitochondrial mutations was established
by Polyak and colleagues.
• These researchers cultured colorectal cancer cells
taken from the tumors of 10 colorectal cancer
patients, and found significant mitochondrial
mutations not present in nearby tissues samples.
• Conclusion: perhaps mutated mitchondrial
enable enhanced ATP production needed by
cancer cells for fast reproduction?
MtDNA accumulates mutations rapidly
• mtDNA accumulates mutations approximately
10 times faster than nuclear DNA.
• Why?:
– repair mechanisms present in the nucleus are
absent in mitochondria
– mitochondria produce oxygen free radicals that
can oxidize DNA and RNA (usually producing
mutations)
– mtDNA lacks histones proteins , which are thought
to protect DNA from damage
The Inner Life of the Cell
The Harvard Cell Video
The XVIVO Version of the Video