bifunctional chemistry (06522)
")
December 2012
BIFUNCTIONAL CHEMISTRY (06522)
Contents Overview
Keto-enol tautomerisation
Reactions of enols: C-Hal and C-N=O bond formation
Reactions of enols and enolates with aldehydes and ketones
Reactions of enolates with acylating agents
Reactions of enolates with alkylating agents
Conjugate additions: the Michael reaction
Reactions of enol and enolate-like molecules
Learning outcomes: At the end of the course you will be able to:
1. Keto-enol tautomerisation
Demonstrate the mechanisms of both acid and base-catalysis of tautomerisation and draw tautomeric form(s) of a given molecule. Discuss factors controlling tautomeric stability.
2. Enols and enolates: C-Hal and C=N-OH bond formation
Work out the products arising from the reaction of enols or enolates with halogens and the
+
NO ion.
3. Enols and enolates: the aldol reaction
Work out the product(s) arising from the reaction of enols or enolates with aldehydes or ketones.
4. Enolates: the Claisen and Dieckmann reactions
Work out the product(s) arising from the reaction of enolates with esters, anhydrides and acid chlorides.
5. Reactions of enolates with alkylating agents
Work out the product(s) arising from the reaction of enolates with alkyl halides.
6.
β-Keto esters and 1,3-diesters
Demonstrate the use of β-keto esters and 1,3-diesters as stable enolate equivalents for such reactions, with the subsequent decarboxylation process.
7.
Reactions of enolates with α,β-unsaturated carbonyl compounds
Work out the product(s) arising from the conjugate addi tion reactions of stable enolates with α,βunsaturated carbonyl compounds.
8. Enamines and Silyl Enol Ethers
Compare the reactions of simple enamines and enol ethers with those of enols and enolates.
9. Nitroalkanes and cyanoalkanes
Compare the reactions of the anions of nitroalkane and cyanoalkanes with those of enolates.
Recommended reading
Organic Chemistry, J. Clayden, N. Greeves, S. Warren and P. Wothers, Oxford University
Press. Chapters 21, 26, 27, 28, 10, 29 (1
17 and 28 (2 nd st Edition) or Chapters 20, 22, 25, 26 and in part
Edition). Background / revision: Chapters 6, 12, 14 (reactions of aldehydes, ketones and esters – lectures from year 1) (1 st Edition) or Chapters 6, 10 and 11 (2 nd
Edition).
Also recommended is Chemistry Of The Carbonyl Group, A Programmed Approach To
Organic Reaction Mechanisms, S. Warren (Wiley, 1974)
Bifunctional Compounds, RS Ward, OP (Oxford Primer No. 17): useful in parts
Similar chapters will be found in all Organic Chemistry texts, e.g. T. Solomons & C. Fryhle,
F. Carey, Maitland Jones etc.
See http://www.hull.ac.uk/php/chsanb/teaching.html for past exam paper questions with model answers. Any comments on or feedback on the usefulness of these notes will be gratefully received. Dr AN Boa, a.n.boa@hull.ac.uk
, 01482 465022.
December 2012
This course is about enols and enolates and their reactions. Each enol or enolate is closely related to a carbonyl-containing compound such as an aldehyde, ketone or ester. Carbonyl compounds may be described as bifunctional as their reactivity as a carbonyl compound is electrophilic whereas the related enol or enolate is nucleophilic. The accompanying PowerPoint slides for this course are split into eleven sections which should be worked through in order.
Follow the self-animated reaction mechanisms by slowly clicking on the forward arrow and paying careful attention to the curly arrows. The slides are supplemented by further explanatory notes in this document which should be used alongside the PowerPoint slides. Make sure you attempt the problems at the end of each section before your progress.
1) Ketones/aldehydes, enols and enolates: Tautomerism (slides 1-7) a.
This section introduces the concept of tautomerism and the equilibrium between ketones/aldehydes and their enol tautomeric form.
2) Reactions of enols and enolates with non-carbon electrophiles (slides 8-10) a.
This section reveals the nucleophilic nature of enols and enolates through their reactions with simple electrophiles.
3) The Aldol reaction (1,3-dioxygenated systems) (slides 11-15) a.
This section introduces the aldol reaction, which is a reaction of an enol or enolate with an aldehyde or ketone. If the enol/enolate is related to the carbonyl compound being attacked then it is referred to as an aldol self-condensation, or if it is different it is an aldol crossed-condensation.
4) The Claisen reaction (1,3-dicarbonyl systems) (slides 16-19) a.
This section introduces the Claisen and Dieckmann reactions, which are reactions of an enolate with an ester. The Dieckmann reaction is a Claisen condensation which forms a ring (sometimes referred to as a Dieckmann cyclisation).
5) Some problems of reactions of enolates (slides 20-22) a.
This section considers some of the problems which can arise in crossed condensations (aldol-type and Claisen-type) when the acidity of the carbonyl compound is low, and/or the reactivity of the electrophile is high. The concept of decarboxylation is introduced which leads into....
6) 1,3-Dicarbonyl compounds (slides 23-24) a.
This section considers the advantages conferred on the reactions of 1,3-dicarbonyl compounds due to the much-enhanced acidity of their α-protons. The alkylation of enolates is considered (electrophile is an alkyl halide).
7) The conjugate addition reaction (1,5-dioxygenated systems) (slides 25-27) a.
This section introduces the conjugate addition (Michael) reaction, which is a reaction of an enolate with an unsaturated carbonyl compound.
8) Acyl anions (slide 28) a.
This section shows how molecules with equivalent reactivity to acyl anions can react to form 1,2- and 1,-4 dioxygenated systems.
9) Enols and enolates: Summary (slides 29-31, and 32 in main handout) a.
This is a short section which summarises the key reactions covered so far, and presents some examples in which more complex products can be made from a sequential application of the reactions covered.
10) Enamines (slides 33-36) a.
This section introduces enamines, and shows comparisons of these nitrogen containing analogues of enols.
11) Nitro- and cyanoalkanes (slides 37-39) a.
The reactions of the anions arising from deprotonation of nitro- and cyanoalkanes are compared to enolates.
December 2012
Explanatory notes and problems to accompany the PowerPoint slides
PowerPoint slides Part 1
1a and 1b
NMR can be used show that certain carbonyl compounds exist in equilibrium with an alternative tautomeric form, the
“enol”. Keto and enol tautomers are isomeric structures, but they not considered as other isomers (e.g. cis and trans alkenes) because they cannot be isolated individually; they are always in equilibrium. Many carbonyl compounds do not show their tautomeric form in the NMR as the enol form is present in too small quantities, but exchangeable protons can be replaced by adding D
2
O. The exchangeable (acidic) Hs are replaced by D and then “disappear” from the NMR spectrum. NB It is important not to confuse enols with alkene-ols as their chemistry is different when the OH is attached directly to the C=C.
2a and 2b
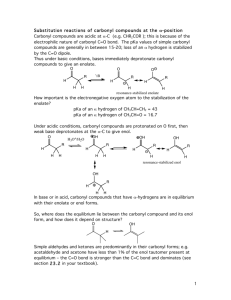
The position of the keto-enol equilibrium depends on the acidity of the CH next door to (or
α to) the C=O (the so called
α–CH, proton on the α–carbon or, misleadingly, the α–proton). In the case of simple ketones and aldehydes the “keto” form (the C=O tautomer whether referring to an aldehyde or ketone) is the more stable and so only the keto form is present in significant quantities. Differences in energies between different tautomeric enols can be seen, and this can influence the course of reactions (see later).
3a, 3b and 4a
The percentage enol in the tautomeric mixture varies greatly depending on the structure of the molecule (the acidity of the α–CH). 1,3-Dicarbonyl compounds contain significant amounts of enol form due to the stabilising features of intramolecular hydrogen bonding and conjugation in the enol form, though solvent will influence this ratio if it is capable of hydrogen bonding itself.
4b and PROBLEMS 1 to 3
1 Identify the ACIDIC HYDROGEN ATOMS in the following molecules by circling. Draw in the relevant H atom(s) first (If necessary); don’t just circle the α-carbon.
O O O
O
H
H
O
O
NC
O
O
O
O
O
O N
O
O
O
O
O
O
O
O
O
O O
O
December 2012
2 Draw all possible ENOLS for the following molecules. Consi der “mono” enols only. If more than one enol is possible, which is the more/most stable and why?
O
O
H
H
O
O
O
O
CO
2
Et
O
O
3 Draw the most stable enol tautomer of each of the four compounds shown below. Assuming the four are present as neat liquids, place them in order of increasing enol content in the tautomeric mixture.
Give a brief explanation for your order.
O O O O O O O
Ph Me
5a and 5b
Some enols are so stable that we consider the enol form as the
“normal” tautomeric structure.
The keto tautomer is, in effect, not present.
6a and 6b
As seen in slide 2b, the ready interconversion of structures helps define them as tautomers as opposed to isomers. However, the rate of interconversion can be accelerated by acid or base catalysis (remember that a catalyst does not affect the position of the equilibrium). In the latter case the interconversion goes via the resonance stabilised enolate. This charged structure is more reactive than the enol due to its negative charge. When considering unsymmetrical ketones the choice of acid or base catalysis may accelerate the interconversion to one enol form over another. In the case of base catalysis start these mechanisms with the lone pair on the base; the arrows then follow one another naturally. In the case of the acid route the C=O lone pair attacks the proton first.
7a and 7b
Tautomerisation, promoted by either acid or base, can result in racemisation or facile double bond migration. Note that in the first example both resonance forms of the enolate are not drawn. It is important to remember however that it is the delocalisation of the charge that contributes to the stability of the enolate. The resonance form with charge on oxygen is much more commonly drawn as a representation of the enolate because the charge is stabilised to greater extent on the electronegative oxygen atom.
December 2012
4c
4d
PROBLEM 4
4a
4b
Draw the mechanism for the FORMATION OF AN ENOLATE from the following compounds. First draw in the acidic α- hydrogen atom if it is not already drawn explicitly.
E.g.
O
EtO
E.g.
EtO
H
O
O
H
2
C CH
3
Add the arrows here
CH
2
H
O
H
2
C CH
3
O
CH
2
O
H
2
C CH
3
O
CH
2
O
O
O
O
EtO
EtO
EtO
PowerPoint slides Part 2
8a and 8b
Acid catalysed halogenation of enols. The enol is nucleophilic and can attack a variety of electrophilic sources of halogen, e.g. molecular bromine or iodine. As the reaction is under acid conditions then the more substituted enol is favoured, and thus the more substituted
α-halo carbonyl compound is produced.
9a and 9b
Under base conditions the less substituted enol is favoured. Also, the initial
α-halo carbonyl compound is more acidic than the starting material and so polyhalogenation can occur easily.
This process forms the basis of the iodoform reaction, which has been used in the past as a chemical test for the presence of a methyl ketone group within a compound.
December 2012
10a and 10b
Enols react with the nitrosonium ion to form α-nitroso carbonyl compounds. These then tautomerise to the more stable oxime which can be reduced to an amine (more later in the heterocycles course and also in Y3).
PowerPoint slides Part 3
11a and 11b
The base-catalysed aldol reaction produces a β-hydroxycarbonyl compound. The examples here are of self-condensations - i.e. where an enolate reacts with its “progenitor” carbonyl compound. Note that the reaction is an equilibrium, i.e. the reverse (or retroaldol) reaction can take place. Thus in the case of unsymmetrical ketones (11b), even where more than one enolate may arise, the steric hindrance may lead to a major product becoming the sole product.
It also means that in cases of unfavourable steric hindrance the amount of β-hydroxycarbonyl compound at equilibrium may be very small (or zero) and the yield of product low (see later).
12a and 12b
The aldol reaction may be catalysed by acid, and may be used to form rings (an intramolecular aldol). This also works with base catalysis. Look out for unsymmetrical dicarbonyl compounds
(12b) in which more than one product may be formed. Unless one enol(ate) is produced specifically then a mixture of products may well be formed.
13a and 13b
The aldol reaction product ( β-hydroxycarbonyl compound) in many cases eliminates water in situ to produce an α,β-unsaturated carbonyl compound – this is particularly favoured when (a) acid catalysis is used, or (b) a more extended conjugated system is produced. As the elimination is essentially irreversible so the problem of the reversibility of the aldol condensation steps is removed. Good yields of α,β-unsaturated carbonyl compounds may be obtained. Note the mechanism of the base catalysed elimination follows the E1cb process. It is a two step elimination (like the E1 elimination) however in the E1cb mechanism the proton is removed first
(fast enolate production), and the slower rate determining step, loss of the hydroxide ion (a poor leaving group), is second.
14a, 14b, 15a and 15b
The crossed aldol condensation: If a mixture of two carbonyl compounds is treated with base then more than one enolate may be produced. Also there will be two electrophiles present (the two carbonyl compounds) and so four products could in principle be made (two crossed condensations and two self condensations). The ideal crossed condensation combination is where one carbonyl compound gives rise to the enolate preferentially, and the other carbonyl compound is a more reactive electrophile. Aromatic aldehydes often are involved in successful crossed condensations as they cannot form enolates (no
α-CH) yet are reactive electrophiles.
Slides 15a and 15b give examples of crossed condensations which are biased to form products selectively, and show how ring size may lead to one cyclisation route being preferred over another.
Some problems associated with the crossed condensation reactions in slides 14 and 15 are found after discussion of the Claisen condensation.
December 2012
5a
5b
PROBLEM 5
Draw the mechanism for the following ALDOL REACTIONS . First draw in the acidic α- hydrogen atom if it is not already drawn explicitly.
E.g.
E.g.
H
O
Add the arrows
O
O
H
H
NaOH
OH
OH
NaOH
NaOH
H
O
O
O
H
O
H
O
O
O
H
O
H
O
H
H
O
HO
H
H
H
O
OH
O
OH
H
H
OH
H
O
H
H
O
OH
O
OH
H
H
O
H
O
O
H
OH
OH
H
O
NaOH
5c
PowerPoint slides Part 4
16a, 16b, 17a and 17b
The Claisen condensation-type mechanism involves esters which have particularly weakly acidic α-CHs ( p K a
~ 25). Thus with “weak” bases, such as ethoxide, only a tiny amount of enolate is produced in the first deprotonation step. However the reaction can be driven to completion by virtue of the product possessing a particularly acidic α-CH (a 1,3-dicarbonyl system with p K a
~ 12). Thus the unfavourable starting deprotonation equilibrium is offset by a favourable one at the end. This also means though that a whole molar equivalent of base is needed as the product enolate formed reacts preferentially with any base left and not the starting ester. Once equilibrium has been reached the alcohol produced (normally methanol or ethanol) is evaporated forcing the equilibrium to shift to the salt of the product. The product is then obtained / isolated by using an acidic work.
The Dieckmann cyclisation is an intramolecular Claisen reaction. In this reaction the two factors which must be considered are (a) which of the two α-CHs next to the ester groups will be deprotonated (or both) and (b) what ring size is formed when the enolate attacks the nearby ester group. The chance of both esters being deprotonated at the same time is very low (see above) and so this situation can in most cases be ignored. Five to seven-membered rings can be made in this way. Smaller rings suffer from strain, and the reversibility of the steps means that the intermediate is likely to collapse back to the enolate-ester. If the ring is too large then either the two ends are too far apart, and “don’t meet”, or else ring transannular effects make the transition states unfavourable.
December 2012
6e
6c
6d
PROBELM 6
6a
6b
Draw the mechanism for the following CLAISEN/DIECKMANN CONDENSATIONS . First draw in the acidic α-hydrogen atom if it is not already drawn explicitly.
E.g.
EtO
E.g.
O
H
H
O
O
OEt
O
OMe
O
OEt
The pKa of ethyl propionoate is about 25, for ethanol it is 16 and for the product it is about 12. Explain why the pKa value of the product is key to the success of the reaction.
Add the arrows
OMe
NaOEt
NaOMe
EtO
O
O
OEt
O
OEt
O
O
EtO
O dil H
3
O
+
O
OEt
EtOH
O
OMe
OEt
O
EtO
O
MeO
O
H
H
O
OMe
OMe
OMe
OMe
OMe
H
OMe
O
MeOH
OMe
O
O dil H
3
O
+
O
O O
O
O
O
O
O
O
O
O
NaOMe
NaOEt
NaOEt
O O
December 2012
18a, 18b, 19a and 19b
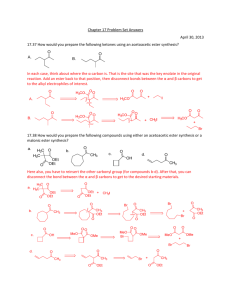
In considering crossed Claisen condensations the same factors need to be considered as in the crossed Aldol condensation. That is (a) how many enolates may be produced, and whether one is formed preferentially over another, and (b) the reactivity of the carbonyl groups as electrophiles. Slides 18a and 18b give some examples of esters which are more reactive than typical aliphatic esters, which do not possess acidic α-CHs and so which preferentially act as the electrophile in a crossed Claisen reaction.
PROBLEMS 7 and 8
– involves crossed aldol and Claisen reactions (look for the electrophiles to distinguish the two mechanism types)
7b
7a
7c
8a
Draw the mechanism for the following CROSSED CONDENSATIONS . First draw in the acidic α- hydrogen atom if it is not already drawn explicitly.
E.g.
O
O
O
NaOEt
O
-EtO
H
O
OEt EtO OEt
O no acidic Hs!
O
O
EtO
O
O
OEt
O
O
H
O
OEt
OEt
O dil H
3
O
+
O
E.g.
Add the arrows
O
O
O
O
OEt
OH
H
O
O
H
O
H
OEt
O O
NaOH
H
Ph
O
H Ph
OH
H
Ph
OH
Ph
O
H no
acidic
Hs!
H
O
H
Ph
OH
H
2 x
EtO
O
Br
CHO
+
EtO
+
O
O
O
OEt
NaOH (aq)
EtOH
NaOEt
Work out all of the possible products that can be obtained when a mixture acetone and butanone are allowed to react together in the presence of aqueous sodium hydroxide (assume elimination of water takes place).
December 2012
8b
8c
O O
O O CH
3
What two compounds could condense to form this molecule?
Is the synthesis from your two starting materials practicable?
What two compounds could condense to form this molecule?
Is the synthesis from your two starting materials practicable?
PowerPoint slides Part 5
20a
Crossed condensations, using the methods discussed before, will not normally work if the enolate wanted has to be generated from the weaker acid, and / or the desired electrophile is less reactive carbonyl compound. In the first example the aldehyde would self condense in preference to either crossed condensation as the
α-CH is more acidic in the aldehyde and the aldehyde is more reactive (than the ester) as an electrophile. It would be useful to be able to react an enolate with other electrophiles, such as alkyl halides. However if the enolate is generated in equilibrium the starting carbonyl compound, then the aldol or Claisen reaction would take place preferentially to (or at least in competition with) the alkylation reaction.
20b, 21a and 21b
A solution to the problems discussed above is to generate the enolate separately, and then add the appropriate electrophile. But in order to do this the enolate must be generated quantitatively and not in equilibrium with the acidic α-CH precursor (or else an aldol or Claisen condensation would take place). The
“quantitative deprotonation” can be done by using a much stronger base; in effect the deprotonation equilibrium lies to the far right.
Lithium bases such as butyl lithium (BuLi) or lithium diisopropylamide (LDA) are very reactive and strong bases (having weak conjugate acids). They must be used under low temperature and anhydrous conditions. The enolates generated cannot self condense and can be reacted by subsequent treatment with a range of carbonyl compounds or alkyl halides.
22a and 22b
An alternative to using a strong and reactive base would be if one could make the starting material more acidic. This can be achieved by using 1,3-dicarbonyl compounds such as 1,3diesters or
β-keto esters as a starting material. The extended conjugation in the enolates of 1,3diesters and β-keto esters means that the enolate is more stable (this is reflected in the lower p K a
of their conjugate acids compared to simple ketones or esters). The “extra” β-ester group can however be removed easily by a sequence of hydrolysis followed by decarboxylation. The latter step is normally spontaneous. Thus the extra
β-ester group can be used to activate (lower acidity of) the α-CH, direct the deprotonation and subsequent reaction, and then afterwards be removed by decarboxylation.
Aromatic acids, such as salicylic acid, will decarboxylate when heated. This acid is in an enolic form of β-keto acid, and heating allows equilibration to the keto form which loses CO
2
.
PowerPoint slides Part 6
23a, 23b, 24a and 24b
These slides give some examples where stable enolates are generated using weak-ish bases
(NaOR) and then reacted with alkyl halides (23a, 24a) or acid chlorides (23b). In the case of
December 2012
9a
9b
1,3-dicarbonyl compounds each free α-CH can be replaced with an alkyl group in either a “one pot” or sequential double alkylation. Also given is an example where the enolate “product” from a Claisen reaction (24b) is reacted directly with an alkyl halide instead of
‘work up’ with an acid.
PROBLEMS 9, 10 and 11
DECARBOXYLATION : Work out the products for the following reactions, identifying any key intermediates in the sequence.
E.g.
O
O
O
CO
2
Me
MeO
2
C
OEt
OEt
CH
3
Ph
NaOH (aq)
1) NaOH (aq)
2) H
3
O
+
, heat
1) NaOH (aq)
O
O
ONa
CH
3
ONa
H
3
O
+ heat
O
H
O
OH
O
CH
3
H
O
O
OH
C O
CH
3
O
OH
CH
3
10c
10a
10b
O
EtO
O
MeO
2
C
2) H
3
O
+
, heat
Draw the mechanism for the following ALKYLATION AND HALOGENATION REACTIONS . First draw in the acidic α- hydrogen atom if it is not already drawn explicitly.
E.g.
HOEt
O O
O O O O
NaOEt
O O
O O O O
H
NaBr
Na OEt
Br
O
O
NaH
PhCH
2
Br
O
O
O
O
OEt
OCH
3
The H in sodium hydride is a strong base causing irreversible formation of enolate [as H
2
(g) produced]
2 mol. equiv. NaH
2 mol. equiv. CH
3
I
NaOEt
CH
2
=CH-CH
2
Br
December 2012
11 Consider the following reaction scheme and answer the following questions.
NaOH
H
2
O
O
O O
OEt
2 moles NaOEt
1 mole
BrCH
2
(CH
2
)
2
CH
2
Br
C
10
H
16
O
3
C
8
H
11
NaO
3
HCl
H
2
O
A B C D
Work out the structure of B and give a mechanism to show how it can be made from A using the reagents given.
Work out the structure of C and show the mechanism of how this is converted to D by warming with aq. HCl.
PowerPoint slides Part 7
25a
Nucleophiles may react with α,β-unsaturated carbonyl compounds at either the carbonyl carbon or the β-carbon. The former is referred to as ‘direct addition’, and the latter as ‘conjugate addition
’. These are also referred to as 1,2- (direct) or 1,4- (conjugate) additions. The origin of this terminology is explained below.
1,2- addition 1,4- addition
Nu
H
O
1
2
R
3
4
1. Nu
-
2. H
+
1 O
R
2
3
4
1. Nu
-
2. H
+
R
2
1
O
H
3
4
Nu R
O
Nu
1. Nu
-
1. Nu
-
2. H
+
2. H
+
Nu O
O
25b and 26a
R R Nu
The types of nucleophiles that prefer conjugate addition reactions can be easily spotted. In this course the nucleophiles considered mainly are stable enolates (such as those from 1,3diketones, esters etc., see 22a) and in these cases the conjugate addition process may be referred to as
‘the Michael reaction’. The stability of the nucleophile also means any initial addition to the C=O is reversible, so whilst 1,2- addition is the faster process it is reversible. So the more stable (retains strong C=O) 1,4-addition product predominates.
With the
α,β-unsaturated carbonyl compounds, those with less reactive carbonyl groups and least steric hindrance at the β-carbon react preferentially in conjugate addition reactions.
26b
Some examples of the Michael reaction are shown here. Note the 1,5 dicarbonyl arrangement in the product which is the signature of a Michael reaction product. The reaction is like the
Claisen reaction in that the acidity of the starting material and product vary greatly, but in this case it is the starting material that is more acidic. This means the reaction could be run using
December 2012 catalytic amounts of base as the product enolate can easily deprotonate the starting material to continue the reaction.
27a and 27b
The Mannich reaction, a reaction of an enol with an iminium salt to produce β-amino ketone, may be coupled to an eliminationin situ conjugate addition sequence. This process allows for a net conjugate addition process without having to make and isolate the
α,β-unsaturated enone
(note the 1,5dicarbonyl ‘signature’ in the product). This is particularly useful if the enone required is reactive.
PowerPoint slides Part 8
28a and 28b
Dithioacetals are ‘protected’ forms of aldehydes (see Dr Eames’ course from Y1). Deprotonated dithioacetals (dithioacetal anions) are therefore equivalents of “acyl anions” and they can be used in addition reactions (both direct and conjugate) to carbonyl compounds to produce 1,2- and 1,4-dioxygenated frameworks.
PowerPoint slides Part 9
29a¸ 29b and 30a
These slides show the use of an approach called retrosynthetic analysis (“paper chemistry”) to examine 1, n oxygenated frameworks and relate them back to pairs of ‘imaginary’ reactants, or synthons , which would react to give the compound being considered. A synthon might look like a familiar reaction intermediate (e.g. an enolate) but strictly the synthon exists alone (i.e. the enolate has an associated counterion). The synthetic equivalent is the reagent(s) which will react in the same fashion as the synthon.
30b, 31a, 31b and 32
These slides show some syntheses in which individual reactions covered earlier in the course can be used sequentially to build up more complicated products. Slide 32 summarises the key reactions that have to be fully understood to succeed in this course.
PROBLEM
Past paper question: 06522 (2009-10)
More past paper questions with worked answers can be found on the following website: http://www.hull.ac.uk/php/chsanb/teaching.html
Answer ALL of the following parts.
O
O O
C
O O
MeO OMe
NaOMe
MeO
O O
NaOMe
MeO
2
C
A B
D
1. NaH
2. PhCH
2
Br
1. aq.NaOH
2. aq HCl, heat
O O
E
C
15
H
18
O
3
F
December 2012
(a) Answer BOTH parts (i) and (ii)
(i) Identify all the acidic hydrogen atoms in F and, giving a brief explanation, draw its most stable enol tautomer.
(ii) Contained within one of the molecules A to F is a hydrogen atom with a p K a
of 13.
Identify this hydrogen atom and the molecule in which it is found.
(4,2 marks)
(b) Answer TWO of the following parts (i) to (iv).
(i) Give a mechanism to show the formation of the enolate of A using sodium methoxide, and then show the subsequent Dieckmann reaction to form B . Explain why a full molar equivalent of base is used in this reaction.
(ii) Compound B reacts with sodium methoxide and methyl vinyl ketone ( C ) to give D .
Show a mechanism for this transformation. Explain why a catalytic amount of base can be used in this reaction.
(iii) Compound B reacts with sodium hydride and benzyl bromide to give E . Identify E and show a mechanism for its formation. Explain why you could not successfully react F with the same reagents.
(iv) Use a mechanism, and show the intermediates, to explain the transformation of D into F .
(2 × 6 marks)
(c) Ketones G and H are readily interconverted upon treatment with base. Rationalise this interconversion.
O
OH
NaOH
G
HO
H O
(7 marks)
PowerPoint slides Part 10
33a and 33b
Keto-enol tautomerism is not the only form of tautomerism (e.g. see nitroso-oxime tautomerism on slide 10a,b). In principle there are many structures which may have tautomeric forms, but only those where the two structures are in equilibrium are considered true tautomers. Imineenamine tautomerism can be considered as the nitrogen equivalent of keto-enol tautomerism.
Remember that imines and enamines can be made from reaction of an aldehyde or ketone with an amine (Dr Eames ‘Chemistry of the carbonyl group’ in Year 1).
34a, 34b, 35a, 35b, 36a and 36b
Silyl enol ethers are semi-stable enol-like molecules which can be made to react like an enol by treatment with an acid or Lewis acid. Thus they are useful for crossed aldol condensations in that the silyl enol ether can be made separately, then a different carbonyl compound added as the electrophilic component for the crossed condensation.
December 2012
Like silyl enol ethers, enamines can also be used as enol equivalents in aldol and alkylation reactions. The enamine group can be hydrolysed with aqueous acid at the end to give back the original ketone making the enamine route another alternative approach for controlling crossed condensations or alkylations. There are some issues in controlling reactions as the enamine can react either at carbon or at nitrogen, This is not a problem with acylation as the initial step is reversible. However with alkylation processes a mixture of products may be obtained depending on the reactivity of the alkyl halide.
Enamines also can be used in conjugate addition reactions and, as with traditional bifunctional chemistry synthetic sequences, these reactions can be run in sequence to form rings. The example on slide 36b shows an enamine conjugate addition followed by an aldol-type process.
The use of enamines was developed by Gilbert Stork in the 50s and 60s. Its popularity dropped off when methods such as the silyl enol ether aldol route were developed. Recent times have seen a resurgence in enamine chemistry by way of the latest vogue topi c: “organocatalysis” which uses chiral amines, such as proline, to make enamines/iminium ions in situ .
PowerPoint slides Part 11
37a, 37b, 38a, 38b, 39a and 39b
The nitro group in organonitro compounds strongly actives/acidifies the
α-CH. For example the p K a
of nitroethane is around 9, about the same as phenol. This means that a nitronate, the anion of a deprotonated organonitro compound, can be made easily using a relatively weak base. Given that nitro compounds cannot self-condensation (the nitro group is not electrophilic like the carbonyl group), these nitronates can be reacted smoothly with aldehydes, ketones, alkyl halides and α,β-unsaturated carbonyl compounds in analogous fashion to those reactions seen with enolates. Many similarities are also seen with organonitriles, where the CN group can also stabilise a negative charge through resonance.
December 2012
1
Extra Problems in Bifunctional Chemistry
You must make sure you complete first the problems in the guidance notes above. The questions below are in the same style, but later questions are harder and the problems may involve one or more consecutive steps (but still using reactions such as the aldol,
Claisen/Dieckmann, conjugate addition etc that have been covered).
Draw all possible ENOLS for the following molecules. Consider “mono” enols only.
If more than one enol is possible, which is the more/most stable and why?
O
2
3
O
O
O
O
O
O
O
Draw a mechanism for the FORMATION OF AN ENOLATE from the following compound.
O
EtO
Draw a mechanism for the following ALDOL REACTION .
O O
NaOH
4 hint: 1,5-dicarbonyl...cyclisation
Draw a mechanism for the following CLAISEN CONDENSATION .
O
O
NaOEt
5
6
7
8
9
December 2012
Draw a mechanism for the following CROSSED CONDENSATION .
O
NaOH
O
NO
2
Draw a mechanism for the following CONJUGATE ADDITION .
NaOEt
O
+
O O
Draw a mechanism for the following ALKYLATION REACTION.
NaOEt
CH
3
I
O
O
DECARBOXYLATION : Work out the products for the following reaction, identifying any key intermediates in the sequence.
Ph
1) NaOH (aq)
EtO
2
C
O
Ph
2) H
3
O
+
, heat
O
Devise a synthesis of the following molecule starting from the compound given, also using the reagents and reactants provided. Hint: do the the previous problem first!
O O starting from
OEt
O O
10
11
O
O
O
O
O and using at some point the following:
NaOH (aq.) NaOEt / EtOH
HCl (aq.)
Br
NaOH (aq.)
NaOMe
C
6
H
8
O
2
C
14
H
20
O
12
13
14
15
16
17
18
19
20
O
O
NaOH (aq.)
O
O
C
13
H
18
O
3
O
O
O
H
2
O
H
+
C
10
H
14
O +
HO
OH
OH
CHO
Cl
K
2
CO
3
O
C
10
H
8
O
2
O
O
O
NaOH (aq.)
O
O
MeO
2
C
EtO
2
C
Me
N
S
CO
2
Me
CO
2
Et
NaH
NaOEt
C
10
H
14
O
2
+
C
7
H
10
O
3
S
C
11
H
17
NO
3
HO
O
CO
2
Me
CO
2
Me
NaOMe
C
8
H
10
O
4
O
O O
O
NaOH
NO
2
C
15
H
27
NO
5
O OEt
NaOEt
C
8
H
14
O
3
+
EtO
O
December 2012
21
22
23
24
O
O
O
O
O
OEt
OEt
O
MeO
O
CO
2
Et
O O
OEt
O
OMe i, K
2
CO
3 ii, H
+
KO t
Bu
MeNH
2
C
11
H
14
O
4
C
9
H
14
O
4
C
7
H
11
NO
3
NaOEt
C
10
H
14
O
4
December 2012