Supplementary information
advertisement

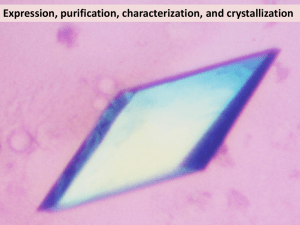
Supplementary Information v410, 235 Supplementary information item 1: MAD phasing PEAK INFLEX REM1 REM2 Wavelength (Å) 0.97923 0.97939 0.99988 0.92873 Resolution (Å) 3.0 3.0 3.0 3.0 # images 300 150 150 150 Total data 253477 125641 125303 132478 Unique data 86086 43136 43258 43177 Completeness(%) 99.8 99.7 99.7 99.8 Mean I/ (I) 16.5 17.7 19.1 12.9 Rmerge (%) Phasing Power (Iso/Anom) 6.6 6.7 5.1 7.6 2.0/2.3 2.5/1.9 0.7/1.5 An inverse beam experiment was performed at the peak wavelength, followed by standard data collection procedures for the remaining wavelengths. Images were collected with an oscillation of =0.5o and an exposure time of 5 seconds. Data were processed using the HKL2000 and CCP4 suite of programs 1-3. A slightly unconventional route was used to estimate Se structure factors. Two Patterson maps were calculated, the first based on anomalous differences (F+peak - F-peak)2, and the second on isomorphous differences (Finflex- Frem1)2. Each was optimised by careful removal of outlying data. Scaling, averaging and back-transformation produced a set of calculated FSe to a resolution of 3.5 Å. Using these data Shake’n’Bake4,5 located 45 of the expected 50 selenium atoms. These were elaborated to 49 atoms and refined using SHARP6. A 3.0Å electron density map revealed 2 molecules in the asymmetric unit and was improved by cyclical 2-fold averaging and solvent flattening (GAP, DIS and JMG unpublished program). An automated model building procedure using BONES7, CA_TRACE (DIS, JMG, unpublished program), CALPHA8 and MUTATE8 generated a model that was rebuilt9 and refined10 against a wavelength-merged data set. 1. Otwinowski, Z. & Minor, W. in Macromolecular Crystallography (eds. Carter Jr., C. W. & Sweet, R. M.) 307-326 (Academic Press, San Diego, 1997). 2. Collaborative Computational Project, N. The CCP4 suite: Programs for Protein Crystallography. Acta Crystallogr. D50, 760-763 (1994). 3. Howell, P. L. & Smith, G. D. Identification of heavy-atom derivatives by normal probability methods. J. Appl. Crystallogr. 25, 81-86 (1992). 4. Weeks, C. M. & Miller, R. The design and implementation of SnB v2.0. J. Appl. Crystallogr. 32, 120-124 (1999). 5. Miller, R., Gallo, S. M., Khalak, H. G. & Weeks, C. M. SnB: crystal structure determination via Shake-and-Bake. J. Appl. Crystallogr. 27, 613-621 (1994). 6. La Fortelle, E. d. & Bricogne, G. in Macromolecular crystallography (eds. Carter Jr., C. W. & Sweet, R. M.) 472-494 (Academic Press, San Diego, 1997). 7. Greer, J. Three-dimensional pattern recognition: an approach to automated interpretation of electron density maps of proteins. J. Mol. Biol. 82, 279-301 (1974). 8. Esnouf, R. M. Polyalanine reconstruction from C positions using the program CALPHA can aid initial phasing of data by molecular replacement procedures. Acta Crystallogr. D53, 665-672 (1997). 9. 10. Jones, T. A., Zou, Y. J., Cowan, S. W. & Kjeldgaard, M. Improved methods for building protein models in electron density and the location of errors in these models. Acta Crystallogr. A47, 110119 (1991). Brunger, A. T. et al. Crystallography and NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Crystallogr. D54, 905-921 (1998). Supplementary information item 2: experimental electron density Sigmaa weighted 2¦Fo¦-¦Fc¦ 2Å resolution electron density for the active site residues D453 and D454, and the octahedrally coordinated Mn++ ion. Supplementary information item 3: 6–HCV sequence alignment Sequence alignment based on structural alignment from SHP 1,2 (see Fig 1b). Strictly conserved residues are in red blocks, and similar residues in blue boxes. Semi-transparent grey bars above the 6 P2 sequence indicate structurally equivalent amino-acids. Secondary structural elements defined by DSSP 3 are shown. The secondary structural elements are coloured by domain with fingers drawn in red, palm in green, thumb in blue, C-terminal domain in yellow and connections between the fingers and thumb in pink. Sequence motifs for RNA polymerases4 are marked by bars below the sequences. Motif A (324-334) is coloured red, motif B (394-413) yellow, motif C (445-461) green, motif D (475-488) blue, motif E (493-505) pink and motif F (268-272) orange. Conserved catalytic aspartates are marked by black ellipses. To avoid breaking up the 6 P2 sequence, residues for HCV polymerase that are not matched by 6 are omitted, and the position and number of residues removed is indicated under the HCV sequence. 1. 2. 3. 4. Stuart, D. I., Levine, M., Muirhead, H. & Stammers, D. K. Crystal structure of cat muscle pyruvate kinase at resolution of 2.6Å. J. Mol. Biol. 134, 109-142 (1979). Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305-8 (1999). Kabsch, W. & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577-2637 (1983). Lesburg, C. A. et al. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6, 937-43 (1999).