Liver/Pancreas Hnd
advertisement

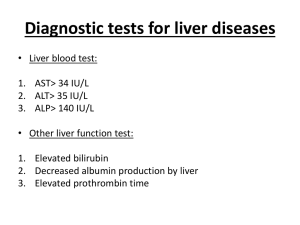
Liver, Biliary Tree & Pancreas Assignment page Reading: Robbins: Chapter 15 (liver) Chapter 16 (pancreas), Wheater: Liver & Pancreas sections Clinical lab Source: - amylase clearance - serum amylase - bilirubin, (know direct & indirect) - HEPATITIS VIRUS antigens & antibodies - AST (SGOT) - ALT (SGPT) - LDH - Alkaline phosphatase - Gamma glutamyl transpeptidase (GGT) - lipase Slides: Liver & pancreas (in the computer image set, the pancreas slides are with the Gastrointestinal section). Please pay special attention to the following: Liver: 4, 5, 18, 42, 46, 47, 63, 69, 71, 103, 115, 132, 137 Pancreas: 75, 92 On-line assignment: There are two cases here, one for liver & one for pancreas. Cases 6 & 7, Mr. Stein & Mr. Weiser. They’re on the web if you chose. These are due by the day of the final exam. I. Pathology C601: Liver, Biliary Tree & Pancreas II. Liver A. Lobule 1. B. C. “Zonation” a) Anatomic b) Physiologic 2. Central vein 3. Sinusoids 4. Canaliculi Triad 1. 3 constituents 2. Limiting plate Cells 1. Hepatocytes 2. Kupffer cells 3. Ductal cells 4. Stellate cells III. General features of hepatic disease /Patterns of injury A. Patterns of injury (As seen by light microscopy) 1. Accumulations a) Fat accumulation (steatosis) b) Protein accumulation (Mallory’s hyaline & alpha-1 antitrypsin) 2. Hepatocyte swelling, necrosis & apoptosis (viral/drug/autoimmune) B. 3. Inflammation 4. Response to injury a) Hepatocyte regeneration/proliferation b) Fibrosis Laboratory assessment of Liver disease 1. Measures of Liver damage (Cytoplasmic hepatocellular enzymes) a) Aspartate aminotransferase (AST) (aka SGOT) (mitochondrial) 2. b) Alanine aminotransferase (ALT) (aka SGPT) (cytoplasm) c) Lactate dehydrogenase (LDH) Biliary tree a) Measures of bile duct damage (Biliary membrane enzymes) b) (1) Serum alkaline phosphatase (Alk Phos) (2) Serum γ-glutamyl transpeptidase (GGT) Substances secreted into bile (1) (2) 3. Serum bilirubin (a) Total: unconjugated plus conjugated (b) Direct: conjugated only Urine bilirubin Measures of Liver function a) Albumin b) Prothrombin (coagulation proteins) c) Ammonia IV. Clinical syndromes A. Hepatic failure 1. Clinical features 2. Acute Hepatitis a) Drug b) Infection c) Microscopic findings in acute hepatitis (1) Hepatocyte injury: swelling (ballooning degeneration), apoptosis (Councilman body) (2) Cholestasis: canalicular bile plugs (3) Hepatocyte necrosis: isolated cells or clusters; If severe: bridging necrosis (portal-portal, central-central, portal-central) 3. (4) Cytolysis (rupture) or apoptosis (shrinkage) (5) Lobular disarray: loss of normal architecture (6) Regenerative changes: hepatocyte proliferation (7) Accumulation of cellular debris in Kupffer cells Chronic Hepatitis a) Some changes shared with acute hepatitis b) Microscopic findings (1) Hepatocyte injury, necrosis, apoptosis, & regeneration (2) Portal tracts: Portal Inflammation , spillover into adjacent parenchyma, necrosis of hepatocytes (“interface hepatitis”), or Bridging inflammation & necrosis. c) Can progress to cirrhosis (1) B. Cirrhosis 1. 2. Causes a) Persistent chronic hepatitis/chronic injury b) Examples (1) Alcoholic liver disease (most common) (2) Postnecrotic cirrhosis (HBV, HCV) (3) Primary biliary cirrhosis (4) Autoimmune hepatitis (5) Metabolic diseases (6) Hemochromatosis (7) Wilson disease (8) α1-Antitrypsin deficiency (9) Galactosemia Microscopic a) Bridging fibrous septa with microvasculature (intrahepatic shunting) b) Nodules c) Diffuse architecture disruption d) 3. Complications associated with cirrhosis a) Progressive Hepatic failure (1) Variable coagulation defects (Increased prothrombin time-PT) (a) Hypocoaguable : ↓synthesis of clot promoting factors (vit K dependent factors –2(prothrombin) 7,9,10) (b) Hypercoaguable: ↓synthesis of anticlotting factors (vit K dependent factors –protein C/S) (2) Hypoalbuminemia – edematous state (a) Pitting edema & ascites - decreased plasma oncotic pressure (3) Hyperestrogenism (a) Males –↓degradation of estrogen & 17-ketosteroids (4) Disrupted urea cycle: ↓ blood urea nitrogen (BUN) & ↑ ammonia (5) Defective gluconeogenesis & decreased glycogen stores (6) Chronic respiratory alkalosis – Toxins overstimulate respiratory center (7) Lactic acidosis (decreased convertion lactic acid to pyruvate) (8) Electrolyte issues - secondary aldosteronism: aldosterone not degraded (9) Vitamin D deficiency - Decreased liver 25hydroxylation of vitamin D (10) Mild ALT/AST elevation. b) Hepatocellular carcinoma c) Hepatic encephalopathy (1) Reversible metabolic disorder (a) ↑ aromatic amino acids (e.g., phenylalanine, tyrosine, tryptophan) (b) Amino acids converted to false neurotransmitters affect glutamine & GABA systems (2) Increased serum ammonia (defective liver urea cycle) (a) Defective Amino acid metabolism in liver (b) Gut flora contribute ammonia (normally broken down) (c) Ammonia (NH3) diffusible, portal vein reabsorption urea cycle (3) (4) (5) Factors precipitating encephalopathy (a) Dietary protein increased (b) Portosystemic shunts - ammonia gets around liver (c) Diuretics Clinical findings (a) Alterations in the mental status including coma (b) Asterixis ( flapping tremor) Treatment: reduce ammonia in the colon (a) Restriction of protein intake, Oral neomycin ( destroy colonic bacteria), Oral lactulose keeps ammonia in the feces d) Portal hypertension (PH) (1) Pathogenesis: Intrasinusoid hypertension, resistance to intrahepatic blood flow, Anastomoses between portal system & arterial system (2) Complications (a) Ascites (i) Increase in portal vein hydrostatic pressure (ii) Hypoalbuminemia - decreases oncotic pressure (iii) Transudate (protein poor)-altered pressures & secondary Aldosteronism (iv) Clinical findings (a) Increased risk for spontaneous bacterial peritonitis (b) Congestive splenomegaly (c) Esophageal varices (d) Hemorrhoids (e) Periumbilical varicies (caput medusa) (3) Therapeutic shunts: Transjugular intrahepatic portosystemic shunt (TIPS) e) (a) Stent connects portal vein with hepatic vein (b) Reduces portal vein pressure (c) Increases risk for encephalopathy Hepatorenal syndrome (1) Intense renal vasoconstriction due to a loss of renal autoregulation C. (2) Reversible renal failure (preserved tubule function ) (3) Shock/Volume depletion/infection (4) 20% of hepatic failure develop ,Mortality >80% Jaundice & Cholestasis 1. Bilirubin & bile formation 2. Functions a) Fat emulsification, bile salt= detergent b) Elimination hydrophobic compounds (bilirubin, excess cholesterol, drugs) and copper 3. Pathophysiology of Jaundice a) Unconjugated Hyperbilirubinemia (1) Blood breakdown- Hemolytic anemias, GI bleed resorption, Ineffective erythropoiesis (2) Reduced hepatic uptake-Drug interference (3) Impaired bilirubin conjugation-Physiologic neonatal jaundice, Breast milk jaundice (4) Genetic deficiency: Crigler-Najjar syndrome, Gilbert syndrome b) Conjugated Hyperbilirubinemia (1) Obstruction – stone, fibrosis, mass (2) Genetic deficiency: Dubin-Johnson syndrome, Rotor syndrome c) Neonatal Jaundice (1) Immature bilirubin conjugation & excretion (< 2 weeks of age) (2) Transient & mild unconjugated hyperbilirubinemia (3) Breastfeeding exacerbates (bilirubin-deconjugating enzymes in breast milk). d) Hereditary Hyperbilirubinemias (bilirubin metabolism defects) (1) Mutations of UDP glucuronosyltransferase (Increased unconjugated bilirubin) (a) Crigler-Najjar syndrome (life threatening) (b) Gilbert syndrome (benign) (2) Deficiency of canalicular membrane transporters (Increased conjugated bilirubin) (a) Dubin-Johnson syndrome ( benign but darkly pigmented liver) (b) 4. Rotor syndrome (benign) Cholestasis a) Impaired bile formation/ flow, accumulation of bile pigment in hepatocyte b) Causes: extra or intrahepatic bile duct obstruction or defective bile secretion c) Clinical: jaundice, pruritus, skin xanthomas (focal accumulation of cholesterol), intestinal malabsorption, including deficiencies of the fat-soluble vitamins A, D, or K d) Laboratory: Elevated alkaline phosphatase & γglutamyl transpeptidase (GGT) e) V. Microscopic. (1) Accumulation of bile pigment in hepatic parenchyma (2) Green-brown plugs of bile within bile ducts. Infectious & inflammatory disorders A. Viral hepatitis (ALT>AST) 1. Infection phases: Incubation Symptomatic, pre-icteric Icteric (jaundice) Convalescence (recovery) 2. Clinical: Jaundice, Fever, Malaise, sometimes hepatic pain 3. Viral hepatitis syndromes a) Asymptomatic infection with recovery b) Symptomatic infection (acute hepatitis) with recovery c) Chronic hepatitis +/- cirrhosis d) Carrier state e) 4. Fulminant hepatitis hepatic failure Death Histologic patterns a) Acute (1) Councilman body (acidophil body), bright pink dead hepatocyte in lobule (2) Inflammation including lymphocytes & Kupffer cell aggregates (3) b) 5. Cholestasis Chronic (1) Increasing fibrosis (2) Lymphocytic inflammation (3) periportal necrosis (aka piecemeal necrosis) Hepatitis A Virus (HAV) a) Incubation: 2-4 weeks b) Mostly subclinical disease & recover; no carrier state; no chronic hepatitis c) Transmission: fecal- oral (eg. Day cares, food handlers, seafood) d) Second most common acute hepatitis in U.S. e) f) 6. Serology (1) IgM –Active infection (2) IgG – recovery/vaccination Hepatitis B Virus (HBV) a) #1 cause of acute hepatitis, #2 cause fulminant hepatitis b) Incubation: 1-4 month c) Variety of outcomes d) e) Transmission: sexual, blood, vertical transmission, breastfeeding (1) Spread via blood (IVDA 50% of cases), accidental needlestick (up to 40% develop HBV; most common mechanism for HBV in health care workers). f) Dane particle = virion g) (1) Envelope protein (Hepatitis B Surface antigen, HBsAg) (2) Core protein (Hepatitis B core antigen, HBcAg) (3) DNA polymerase/reverse transcriptase (Hepatitis B e antigen, HBeAg) (a) h) HBeAg indicator of viral replication, infectivity Antibodies (1) Anti-HBs antibody (IgG) end of acute disease, protective - vaccination (2) IgM anti-HBc (a) Detectable with elevated serum aminotransferases (b) Detectable during serologic gap (c) i) 7. Serology Hepatitis C Virus (HCV) a) Transmission: Blood; #1 hepatitis due to IV drug abuse (70% of cases) (1) Other causes: Transfusions before 1987, accidental needlestick (5% chance of HCV), tattoo. b) #1 indication for liver transplantation. c) Chronic hepatitis in 85% of untreated cases; 20% develop cirrhosis d) Complications: hepatocellular carcinoma (HCC) (2% risk/ year) VI. a) Detection (1) Anti-HCV IgG indicates active infection or recovery (Not protective) b) Confirmatory tests (1) HCV RNA detection c) Note: Sofosbuvir nucleotide analog released 12/2013, cures 90 percent of HCV + patients in 3-6 months, although expensive ($1000 / pill) 2. Hepatitis D Virus (HDV) a) Defective virus - requires HBV virus to replicate b) 2 mechanisms (1) Coinfection: HBV & HDV acute hepatitis is usually self-limited. (2) Superinfection: in chronic HBsAg carrier, severe acute hepatitis, 80% progress to chronic hepatitis, risk of cirrhosis & hepatocellular cancer (HCC). c) Detection (1) IgM anti-HDV indicator of recent HDV exposure(no protective antibody) 3. Hepatitis E Virus (HEV) a) Enteric/water-borne zoonotic disease, animal reservoirs. b) Young to middle age c) high mortality rate amongst pregnant women d) IgG is protective 4. Hepatitis G Virus (HGV) 5. Bacterial, Other viral, Parasitic, Helminthic, infections a) Bacteria - Leptospira b) Other Viral (1) Epstein Barr virus (EBV) Infectious mononucleosis (2) Cytomegalovirus( CMV)- neonatal hepatitis & immunosuppressed (3) Yellow fever (flavivirus) c) Parasites/Helminths –Echinococcus/Liver flukes (Shistosoma Clonorchis) B. Autoimmune hepatitis 1. Young women 2. Associated with HLA- DR3 & DR4 3. Other autoimmune associations: eg. Lupus, Hashimoto thyroiditis 4. Clinical: Fever, jaundice, hepatosplenomegaly 5. Increased transaminases, fulminant hepatitis cirrhosis 6. Chronic hepatitis with clusters of plasma cells in portal areas 7. Laboratory findings a) Positive serum antinuclear antibody (ANA, >60% of cases) b) Anti–smooth muscle antibodies (SMA, >85% of cases) c) Anti-Liver & Kidney microsomal (ALKM) C. Pyogenic liver abcesses -Bacterial or parasitic VII. Alcohol & toxin induced disease A. Alcoholic liver disease (ASH) (AST>ALT) (mitochondrial enzyme predominance) 1. Three types of disease: fatty change, hepatitis, cirrhosis 2. Fatty change most common a) Shunt substrates toward lipid biosynthesis (excess reduced NADH +) 3. Mechanism unknown, contributing factors a) Acetaldehyde induces lipid peroxidation, disrupt cytoskeleton & membrane function, Induction of P-450 radicals 4. Pathogenesis a) Acetaldehyde damage to hepatocytes/mitochondria b) 5. Collagen deposition (fibrosis) Microscopic findings a) Fatty change with Neutrophils! b) Mallory bodies - Damaged cytokeratin intermediate filaments in hepatocytes c) 6. Perivenular fibrosis cirrhosis Clinical findings a) Painful hepatomegaly, Fever, ascites, hepatic encephalopathy 7. b) 15 % progress to alcoholic cirrhosis c) Bleeding from inadequate coagulation proteins (1) Vitamin K deficiency (2) Prothrombin & other coagulation factors Laboratory findings a) Neutrophilia (↑ neutrophil count in peripheral blood) b) Serum AST > ALT (Alcohol damages mitochondria) 8. Info: 80 gm of alcohol (six beers) over span of days mild, reversible steatosis B. Other toxin/drug induced liver disease 1. Serologic markers of viral infection only distinguishing factor 2. Different types of injury a) Cholestasis: Contraceptives, estrogen, antibiotics b) Hepatocyte necrosis/ fulminant hepatic failure: #1 cause is acetominophen C. Special pediatric conditions 1. Reye syndrome a) Association with aspirin + infection (chickenpox, influenza) b) Postinfectious triad: Encephalopathy, Microvesicular fatty change, Transaminase elevation (AST/ALT) c) Affects liver & brain (1) Mitochondrial damage (? virus, salicylates) (2) Disrupts urea cycle (metabolize ammonia), increase serum ammonia (3) Defective β-oxidation of fatty acids, Microvesicular fatty change VIII. Metabolic Liver disease A. Nonalcoholic fatty liver disease (NAFLD) (ALT>AST) 1. 2. Hepatic steatosis (fatty liver), (similar to alcoholic steatosis) a) #1 cause of chronic liver disease in the United States b) Strong association with obesity Includes nonalcoholic steatohepatitis (NASH) a) Hepatocyte injury cirrhosis 10% - 20% b) Microscopic: hepatocyte ballooning, lobular inflammation, & steatosis. c) So called “cryptogenic cirrhosis” 3. AST/ALT ratio < 1 (contrast alcoholic steatohepatitis AST/ALT > 2.0) IX. Inherited liver disease A. Hemochromatosis 1. Autosomal recessive, chromosome 6, HLA-A3 association 2. Most common genetic disorder in North European ancestry 3. Fifth decade, more common in men 4. Normal function (1) HFE protein aids transferrin at duodenum receptors. (2) Hepcidin: iron regulatory hormone produced in Liver, inhibits iron absorption in gut (3) ↓ transferrin-bound iron (↓ iron stores) ↓ synthesis of hepcidin ↑ duodenal reabsorption of iron. 5. Pathogenesis a) Mutated: hereditary hemochromatosis gene (HFE), transferrin receptor or hepcidin (1) HFE: Two missense mutations (C282Y & H63D) (2) Mutated HFE prevents interaction with hepcidin → Unrestricted reabsorption of iron in small intestine, abundant iron produces hydroxyl free radicals fibrosis b) Multorgan iron deposits: “Bronze diabetes” (1) Liver: Cirrhosis (60% of cases) - ↑ risk of hepatocellular carcinoma (2) Pancreas (a) Type I diabetes mellitus (60% of cases) Destruction of β-islet cells (b) Malabsorption - Destruction of exocrine pancreas (3) Skin: Hyperpigmentation -Iron deposits increase melanin production (4) Heart: Restrictive cardiomyopathy (5) Pituitary: Hypogonadism (6) Joints: Degenerative joint disease (>40% of cases) Chondrocalcinosis c) Laboratory findings (1) ↑serum iron, % saturation, ferritin; ↓Total iron binding capacity (TIBC) (2) Transferrin levels (surrogate marker for TIBC) best screening test. B. Wilson Disease (hepatolenticular degeneration) 1. Autosomal recessive, 3-40 years (Usually late childhood) 2. Copper (Cu) normal metabolism a) Ceruloplasmin - Protein synthesized by liver holds six copper atoms (1) Secreted into plasma = 95% of total serum copper. (2) Degraded by liver, copper excreted into bile. 3. Pathogenesis: copper accumulation & multiorgan damage a) Gene defect - affects copper transport (1) Decreased copper incorporation into ceruloplasmin (decreased ceruloplasmin) (2) Decreased biliary copper excretion (Liver copper accumulation). b) Unbound copper attaches to albumin, increases hydroxyl radicals c) Unbound copper spills into circulation (increased blood/ urine copper) damages brain, kidneys, cornea, & other tissues. 4. Clinical findings a) Kayser-Fleischer ring (~70% of cases) (1) Copper deposits in corneal Descemet membrane (also see in primary biliary cirrhosis) from blocked copper excretion b) Intrahepatic copper (1) Hepatospenomegaly (2) Acute hepatitis to cirrhosis. c) Central nervous system disease copper deposition (lentiform nucleau) (1) Parkinsonism, hemiballismus, dementia = lenticular degeneration 5. d) Hemolytic anemia e) Renal disease Laboratory findings a) Early: Decreased serum ceruloplasmin b) C. Late; Increased serum & urine free copper ɑ-1 antitrypsin (α1AT) deficiency 1. Function: α1AT protects tissues (eg. Lung) from enzymes of inflammatory cells, especially neutrophil elastase 2. Mechanism: Decreased α1AT levels in serum, Lack of circulating α1AT allows panacinar emphysema to develop, Accumulation α1AT in hepatocytes (cannot excrete) causes liver damage 3. Autosomal dominant (codominant) 4. Alleles & genotypes a) Normal phenotype (1) Normal allele is M (95% frequency in the United States). (2) PiMM is normal genotype with α1AT in normal range. b) Deficiency- Liver defect in excreting α1AT (accumulates in liver) (1) Deficient variant Z allele (1% frequency) (PiZZ) (2) Deficient variant S allele (2%–3% frequency) (PiSS) c) PiMZ heterozygous genotype has decreased production d) PiZZ homozygous genotype most severe deficiency e) Periodic acid–Schiff (PAS) stain red cytoplasm granules (accumulating α1AT ) on microscopic section X. f) Increased risk for hepatocellular carcinoma g) Most common cause of cirrhosis in children. Disease of the biliary tract A. Neonatal Cholestasis: 1. Multiple etiologies Eg. Maldevelopment of biliary tree (Atresia) 2. Variable prognoses: eg. Untreated Atresia, results in liver failure, cirrhosis by several months of age, and death by 1 yr of age B. Primary Biliary Cirrhosis (PBC) 1. Autoimmune disorder granulomatous destruction of bile ducts lead to cirrhosis 2. Women (>90% ) 40- 50 years old 3. Association with other autoimmune diseases (e.g.,Lupus, Sjögren syndrome) 4. Complications: Increased risk for hepatocellular carcinoma 5. Pathogenesis a) Unknown mechanism: possible environmental insult affects mitochondria triggers CD8 T-cell destruction of bile duct epithelium. b) Antimitochondrial antibodies develop against the mitochondria. 6. Clinical findings a) Early: Pruritus, well before jaundice appears, Painful hepatosplenomegaly b) Late: Jaundice, after most bile ducts have been destroyed c) Other findings: (1) Inflammatory arthropathy (2) Xanthelasma: cholesterol from bile backs up into the blood (3) Kayser-Fleischer rings from copper retention (copper eliminated in bile) 7. Laboratory findings a) Serum ANA positive b) Antimitochondrial antibodies positive c) Increased: serum IgM, ALk. Phosphatase, GGT, Cholesterol 8. Diagnosis ; Biopsy Inflammation of the bile ducts, characterized by intraepithelial lymphocytes, and Periductal epithelioid granulomata 9. C. Secondary biliary cirrhosis (eg cystic fibrosis) Primary Sclerosing Cholangitis (PSC) 1. Obliterative fibrosis of intrahepatic & extrahepatic bile ducts 2. Genetic predisposition: (HLA-DR52a & HLA-Cw7 ) 3. Male dominant (70% ); <45 years old 4. Associations a) Inflammatory bowel disease (70%), Ulcerative colitis > Crohn disease 5. 6. Complications a) Cirrhosis b) Cholangiocarcinoma Clinical findings a) Pruritus - Deposition of bile salts in skin b) Jaundice c) Hepatosplenomegaly 7. 8. Laboratory findings a) Bilirubinuria with conjugated Bilirubin >50% b) GI) Absent urine Urobilogen (obstruction of excretion into c) Increased: serum ALk Phosphatase & GGT Diagnosis a) Endoscopic retrograde cholangiopancreatography (ERCP)-Dye study shows “beading” = narrowing & dilation of bile duct b) Biopsy findings: periductular fibrosis “Onion- skin” D. Other intrahepatic biliary tree anomalies (see bile duct section XIII) XI. Circulatory Disorders of the Liver A. Impaired Blood flow to liver (prehepatic obstruction) 1. Hepatic artery thrombosis with infarction -Uncommon due to dual blood supply. a) 2. Causes (1) Liver transplant rejection (2) Vasculitis due to polyarteritis nodosa (PAN) Portal vein thrombosis a) Causes (1) Pylephlebitis (inflammation of portal vein) - often from bacteria in the portal vein secondary to acute appendicitis (2) Polycythemia vera (hyperviscosity of blood) (3) Hepatocellular or other carcinoma- Tumor invasion of the portal vein b) B. Clinical findings (1) Portal hypertension, ascites, splenomegaly (2) No hepatomegaly Impaired blood flow through liver (Intrahepatic obstruction) 1. Causes a) Cirrhosis (see appropriate section) b) Sickle cell disease c) Centrilobular hemorrhagic necrosis (from heart failure) (1) (2) Left-sided heart failure (LHF) (a) Decreased cardiac output, causes liver hypoperfusion (b) Leads to necrosis of zone III (pericentral) hepatocytes Right-sided heart failure (RHF) (a) Venous regurgitation into central veins, sinusoids, portal vein (b) (3) Enlarged mottled red liver (“nutmeg” liver) Clinical findings: Painful hepatomegaly +/- jaundice, May progress to cardiac cirrhosis (pericentral fibrosis) (4) Lab values: Increased serum transaminases from ischemic necrosis (5) Biopsy microscopic: Congestion of central veins & sinusoids with pericentral necrosis d) Peliosis hepatis (sinusoidal dilation due to blood congestion) (1) (2) C. Causes (a) Anabolic steroids (b) Bacterial infection (angiomatosis) (seen in AIDS) Potential for intraperitoneal hemorrhage Hepatic venous outflow obstruction ( post hepatic obstruction) 1. Causes a) Sinusoidal obstructive Syndrome (SOS)(formerly Venoocclusive disease VOD) (1) Complication of drugs & bone marrow (BM) transplantation (2) b) Collagen develops around & within central venules. Hepatic vein thrombosis (Budd-Chiari syndrome) (1) Causes (a) Polycythemia vera (due to hyperviscosity) (Most common) (2) Hypercoagable state (20% of cases) (a) Oral contraceptive pills (b) Coagulation disease: Proteins C & S deficiency, Antiphospholipid syndrome c) Hepatocellular or other carcinoma- Tumor invasion hepatic vein 2. Clinical findings: Painful hepatosplenomegaly, Portal hypertension, ascites 3. Emergency: High mortality rate (75% die in 1 year) 4. Laboratory findings a) Increased serum transaminases b) Increased Prothrombin time (sign of liver failure) 5. Diagnosis: Ultrasound with Doppler ( first-line), MRI 6. Treatment: Anticoagulation/thrombolysis/stent XII. Bone marrow transplantation (BMT) A. Graft-versus-host disease (GVHD) 1. Background: After bone marrow transplantation to treat myeloma or some leukemia, T cells present in the graft, attack the host tissues (eg. Liver) 2. Acute GVHD (10 to 50 days after BMT): Donor lymphocytes (chronic inflammation) attack liver bile ducts 3. Chronic GVHD (> 100 days after BMT): Endothelitis, Portal tract inflammation, bile ductopenia (loss from destruction), fibrosis. 4. Cholestasis may be observed in both acute & chronic GVHD. XIII. Pregnancy related Hepatic disease A. Acute Fatty Liver of Pregnancy 1. Abnormality in fatty acid β-oxidation in mother, from and induced deficiency of a specific fatty acid enzyme 2. B. Can be fatal to mother & fetus unless the baby is delivered Preeclampsia & eclampsia 1. Hypertension, proteinuria, dependent pitting edema in third trimester 2. Periportal hepatocyte necrosis (zone 1), fibrin deposits hematoma rupture 3. Increased serum transaminases 4. Eclampsia = Preeclampsia + hyperreflexia & convulsions (CNS symptoms) 5. HELLP syndrome a) 2O to disseminated intravascular coagulation (DIC) b) HELLP: Hemolytic anemia, ELevated serum transaminases, Low Platelets C. Intrahepatic cholestasis of pregnancy 1. Pruritus in the third trimester 2. Darkened urine & +/- light stools & jaundice 3. Serum bilirubin (mostly conjugated) mild elevation XIV. Masses & mass-like disease A. Causes: Non-neoplastic nodules, benign/malignant neoplasms, non-neoplastic nodules, abscesses, parasites B. Focal nodular hyperplasia (FNH) 1. Benign incidental non-neoplastic nodule 2. Women > men, possibly estrogen responsive 3. CT scan: Hypervascular mass with arteriovenous connections 4. Gross/micro: Poorly encapsulated nodule, Central stellate scar, large vessels C. Benign tumors of the liver 1. Cavernous hemangioma a) Vascular tumor, most common benign liver tumor b) Risk for intraperitoneal hemorrhage c) Best characterized by CT scan 2. Liver (hepatic) adenoma a) Benign tumor of hepatocytes & vessels (no bile ducts) b) Women > men c) Cause: hormone therapy: Oral contraceptive (most common cause), Anabolic steroids (regress with withdrawal) d) D. Highly vascular, Tendency to rupture during pregnancy Malignant tumors of the liver 1. Metastasis - Most common type of cancer involving liver, usually multiple masses a) 2. Primary lung> gastrointestinal> breast/ kidney Hepatocellular carcinoma (HCC) a) Most common primary liver cancer b) Males > females, Peak age 50-60 yrs c) Causes (1) Most common: Preexisting HBV/HCV cirrhosis (2) Other cirrhotic diseases: Alcoholic cirrhosis, hemochromatosis, Wilson disease , αAT deficiency, Primary biliary cirrhosis (PBC) (3) Environmental: Aflatoxins (from Aspergillus mold in grains & peanuts) d) Gross findings (1) Focal or diffusely infiltrating cancer near preexisting cirrhosis (2) Portal & hepatic vein invasion is common. e) Microscopic findings: tumor pseudoglands arranged with central bile . f) Clinical findings (1) Most have rapid enlargement of the liver in the face of cirrhosis. (2) g) Most common metastasizes to lung Laboratory findings (1) Increased α-fetoprotein (AFP), Sudden increased alk phosphatase & GGT (2) Rarely produce ectopic hormones: Erythropoietin (EPO; secondary polycythemia), Insulin-like factor (hypoglycemia), Parathyroid hormone -related (PTHr) protein (hypercalcemia) h) 3. Prognosis: If resectable, 5-year survival ~40% Angiosarcoma a) Malignant vascular tumor b) Exposure to vinyl chloride (PVC pipes) (most common), arsenic, or thorium (thorotrast dye – old x-ray contrast media) 4. Cholangiocarcinoma (see extrahepatic cholangiocarcinoma) 5. Hepatoblastoma a) Rare malignant tumor of children (1 in 1 million) b) Two anatomic variants: (1) Epithelial type: small fetal/embryonal cells forming acini, tubules & papillary structures (recapitulating liver development) (picture) (2) Mixed epithelial & mesenchymal type, foci of mesenchymal differentiation - primitive bone, cartilage, or striated muscle XV. Biliary Tract A. Cystic diseases of the biliary tract 1. Choledochal cyst a) 10 Congenital dilation of common bile duct, presents < age b) Clinical findings (1) Biliary colic, abdominal pain +/- jaundice (2) Increased incidence of cholelithiasis, cholangiocarcinoma, & cirrhosis c) Ultrasound best screening d) Treatment: surgery. 2. Other cystic biliary diseases (can be associated with autosomal dominant polycystic kidney disease) a) Von Meyenburg Complexes(bile duct hamartoma) (1) Small dilated bile duct clusters & fibrous stroma within portal tracts (2) Common, benign, mimics metastases to the liver in imaging studies (3) b) Multiple can indicate polycystic disease of the liver Polycystic Liver Disease (1) Multiple diffuse cysts lined by cuboidal or flattened biliary epithelium, contain straw-colored fluid c) (1) Congenital Hepatic Fibrosis Persistence of the embryonic biliary tree (2) Portal tracts enlarged with broad b&s of fibrous collagen & abnormal bile ducts causing portal hypertension (bleeding varices) d) Caroli disease (1) Segmental dilatation of intrahepatic bile ducts & portal tract fibrosis (2) Autosomal dominant & recessive types (3) Clinical findings (a) ↑risk for cholangiocarcinoma, cholangitis, portal hypertension, intrahepatic stones/abscesses (4) 3. Treatment is surgery/transplant Cholangiocarcinoma a) #1 malignancy of bile ducts b) Klatskin tumor – Cholangiocarcinoma at Liver hilum, cause jaundice early c) Causes: (1) Primary sclerosing cholangitis (#1 cause in United States) (2) Clonorchis sinensis (Chinese liver fluke) (3) (4) d) e) Thorotrast (thorium dioxide) Choledochal cyst, Caroli disease Locations (1) Ampulla or common bile duct (most common) (2) Junction of right/left hepatic duct (Klatskin tumor) (3) Intrahepatic Clinical findings (1) Obstructive jaundice (2) Palpable gallbladder (Courvoisier sign) (3) Hepatomegaly f) Microscopic: Adenocarcinoma with prominent sclerosis B. g) Diagnosis: Ultrasound, ERCP h) Treated surgically Gallstones (cholelithiasis) 1. Bile: bile salts/acid (~70%), phospholipid(~20%), Remaining: protein, conjugated bilirubin, free cholesterol, bicarbonate 2. Excrete ~ 1 liter/day 3. Water soluble/Detergent renders cholesterol soluble in bile 4. Stone Types a) Cholesterol stones (75%, yellow) mostly radiolucent, only radiopaque if contain calcium carbonate b) Pigment stones (Black pigment stones (Calcium bilirubinate) (1) Extravascular hemolytic anemia (e.g. sickle cell disease), ↑ Unconjugated Bilirubin (from RBC destruction) + calcium → calcium bilirubinate stones c) Brown pigment stones - Sign of infection in common bile duct 5. Pathogenesis of Cholesterol stones a) Supersaturation of bile with cholesterol or decreased bile salts/acids/Phospholipid (Bile salts & acids normally solubilize Cholesterol in bile.) b) Risk factors: Fat Female Fertile Forty c) Estrogen increases cholesterol stones: increases HDL ( transports cholesterol), causes increased LDL uptake, & increases HMG-CoA reductase activity (more cholesterol is synthesized). d) Native Americans at increased risk (e.g., Pima & Navajo Indians) 6. Complications associated with stones a) Cholecystitis (most common)-inflamed gallbladder b) Bile Duct obstruction, ascending cholangitis, carcinoma, acute pancreatitis C. Acute cholecystitis 1. Women > men, 50-60 year olds; High incidence in Native Americans 2. Associated with gallstones in >95% of cases 3. Stages of development of acute cholecystitis a) Stage 1: Stone lodges in cystic duct Midepigastric colicky pain occurs from contraction b) Stage2: Stone impacts cystic duct pain shift to RUQ; radiation to right scapula/shoulder via Chemical irritation of mucosa, Bacterial overgrowth (E coli) c) Stage 3: bacterial invasion GB wall; + Murphy sign; subsides if stone falls out d) 4. Stage 4: perforation, gangrenous necrosis Clinical: Fever, Vomiting, Murphy’s sign (pain on palpation) a) Jaundice indicates a stone in the Common Bile Duct (choledocholithiasis) 5. Laboratory findings a) Neutrophilic leukocytosis with left shift b) Increased Alkaline phosphatase & GGT (Gammaglutamyl transpeptidase) c) Increased amylase suggests associated pancreatitis 6. Ultrasound (US) is the best screening test for stones (Only 20% are radiopaque), shadowing of stone 7. Hepatobiliary iminodiacetic acid (HIDA) radionuclide scan: Stone present if no tracer in the duodenum 8. Treatment a) Cholecystectomy (laparoscope preferred) b) ERCP with sphincterotomy(of Oddi) to extract the stone in the CBD D. Chronic cholecystitis 1. Most common symptomatic disorder of Gallbladder 2. Cholelithiasis with repeated attacks of minor inflammation 3. Severe, persistent pain 12 hours postprandially in the evening E. Cholesterolosis: excess cholesterol in bile; speckled yellow mucosal surface F. Hydrops of the gallbladder: chronic obstruction of cystic duct 1. Distended gallbladder with atrophy of the mucosa/muscle with clear secretions G. Ascending cholangitis 1. Bile duct inflammation (cholangitis) from synchronous biliary infection & duct obstruction (e.g., stone), causes liver abcesses H. 2. Life-threatening disease, usually Escherichia coli 3. Charcot’s Triad: Fever, Jaundice, RUQ pain. Gallbladder adenocarcinoma 1. Elderly women-Poor prognosis (5 yr survival <2%) 2. Pathogenesis a) Chronic Cholelithiasis (95% of cases) b) Porcelain gallbladder - Gallbladder diffusely replaced by dystrophic calcifications c) Approximately 50% risk for progression to cancer XVI. Pancreatic Pathology I. Exocrine Pancreatic Disorders (We deal with exocrine pancreas here with some endocrine, the majority of the endocrine portion will be dealt with in the endocrine unit of the course next semester in diabetes unit) A. Embryologic abnormalities of the pancreas 1. Annular pancreas (malrotation of Dorsal and Ventral pancreas buds) a) Dorsal & ventral buds form a ring around the duodenum. b) Associated with small bowel obstruction. 2. Heterotopic pancreatic tissue (aka. Choristoma/heterotopic rest) a) Locations: Stomach wall (commonly), small intestine or Meckel diverticulum (1) B. Acute pancreatitis 1. Alcohol abuse & gallstones (Choledocholithiasis) major causes. 2. Mechanisms: a) Components (1) Trypsin: important activator of proenzymes (proteases, lipase, lipase) (2) Activated Proteases damage acinar cell structure (3) Activated Lipases & phospholipases enzymatic fat necrosis b) Obstruction of the main pancreatic duct or terminal common bile duct (CBD) (1) use Gallstones or thickened ductal secretions via alcohol (2) Increased back pressure causes bile to reflux into the major pancreatic duct Activation of proenzymes via trypsin pancreas autodigestion c) Acinar cell injury (1) Chemical injury (eg. alcohol, triglycerides) (2) Infectious injury (CMV, mumps, coxsackievirus) (3) Mechanical injury (seat belt trauma(kids), penetration of duodenal ulcer) d) Metabolic activation of proenzymes (e.g., hypercalcemia, ischemia, shock) 3. Clinical findings a) Fever, nausea, & vomiting, knife-like epigastric pain radiating to back b) Hypovolemic shock due to “third spacing” (1) fluid unavailable for volume maintenance in vascular compartment (eg. peripancreatic collection of fluid that commonly occurs as pancreas autodigests) c) Hypoxemia/ARDS in acute pancreatitis: circulating phospholipase destroys pulmonary surfactant d) Physical exam: Grey-Turner sign (flank hemorrhage), Cullen sign (periumbilical hemorrhage) e) Disseminated intravascular coagulation (DIC), prothrombin activation f) Tetany: Hypocalcemia is caused by enzymatic fat necrosis. 4. Complications a) Pancreatic necrosis, Peripancreatic infections, Pancreatic abscess b) Pancreatic pseudocyst (20% of cases) Collection of digested pancreatic tissue around the pancreas ( third spacing) c) Presents as an abdominal mass with elevated serum amylase >10 days d) Treatment: Self-resolving or drainage 5. Prognosis: Full-blown acute pancreatitis is a medical emergency, “Ranson criteria” are rules for predicting the severity of acute pancreatitis. 6. Laboratory: a) Serum amylase (1) Elevated in Acute Pancreatitis: Returns to normal in 2 to 4 days (2) Present in urine (3) Salivary amylase also present that can cause false elevation (a) Salivary amylase increased in mumps (b) Alcoholics often have elevation of salivary amylase without pancreatitis (c) Macroamylasemia – immune complexed amylase, acquired especially with autoimmune disorders, Appears as elevated amylase without symptoms of pancreatitis b) c) Serum lipase (1) Elevated in Acute pancreatitis (2) Not elevated with mumps or Macroamylasemia Newer marker Serum immunoreactive trypsin (1) Specific for the pancreas. (2) Excellent newborn screen for cystic fibrosis (CF increased) (3) Elevated with acute pancreatitis, pancreatic adenocarcinoma, decreased in chronic pancreatitis d) Neutrophilic leukocytosis e) Hypocalcemia, hyperglycemia (destruction of β-islet cells) 7. Imaging studies a) CT scan is the gold standard for pancreatic imaging. 8. Treatment - NPO until clinically improved, Total parenteral nutrition (TPN) C. Chronic pancreatitis 1. Men>women 2. Majority idiopathic, known causes: a) Alcohol abuse is the most common known cause overall. b) Cystic fibrosis is the most common cause in children. c) Malnutrition is the most common cause in developing countries. d) Autoimmune pancreatitis responds to treatment with corticosteroids, regarded as a form of hyper-IgG4 disease (disease in which inflammatory cells cause fibrosis) 3. Pathogenesis: Repeated attacks of acute pancreatitis produces duct obstruction. 4. Clinical findings: a) Severe pain radiating into the back b) Malabsorption - Indicates >90% exocrine function destroyed (Steatoorrhea) 5. 6. c) Type 1 diabetes mellitus (70% of cases) d) Pancreatic pseudocyst Laboratory a) Unreliable amylase & lipase b) Decreased serum immunoreactive trypsin Tests for pancreatic insufficiency a) Secretin stimulation test: Tests ability of pancreas to secrete fluids b) Microscopic: fibrosis and acinar atrophy D. Cysts/Neoplasms Exocrine Pancreas 1. Cysts – Majority of cysts are pseudocysts only ~10% are neoplsms 2. Serous Cystadenoma a) Women > Men, 70 yr old b) Benign neoplasm, 25% of all pancreatic cystic neoplasms, no cancer risk c) Gross: Sponge-like microcysts with straw colored fluid d) Microscopic: Cuboidal cells surrounding small cysts e) Clinical: vague abdominal pain f) Genetics: Mutation of von Hippel-Lindau (VHL) tumor suppressor gene 3. Mucinous Cysts – both have risk for associated Pancreatic adenocarcinoma a) Mucinous cystadenoma (1) Mucin-producing neoplasm, <20% chance of associated carcinoma (2) 95% in perimenopausal women (3) Location: body or tail of the pancreas, not connected to ducts (4) Gross/microscopic: Cyst filled with thick mucin & lined by a columnar mucin-producing epithelium with an associated dense ovarian-type stroma similar to ovarian stroma (5) Diagnosis: CT scan, Ultrasound guided endoscopic fine needle aspiration. Muc b) Intraductal papillary mucinous neoplasms (IPMNs) (1) Mucin-producing intraductal neoplasm, 50% chance of associated carcinoma (2) Men>>women, 70-80 year olds (3) Location: head of the pancreas, within pancreatic duct (4) Gross/microscopic: Cyst filled with thick mucin & lined by a columnar mucin-producing epithelium with lack the dense “ovarian” stroma seen in mucinous cystic neoplasms, & involve a larger pancreatic duct (5) Diagnosis: CT scan, Ultrasound guided endoscopic fine needle aspiration. 4. Pancreatic adenocarcinoma (Exocrine pancreatic cancer) a) Men > women, 70-80 yr old, can occur earlier b) Predisposing factors c) (1) Tobacco use & Smoking (#1 cause) (2) Chronic pancreatitis from any etiology (3) High saturated fat diet; obesity; cirrhosis Pathogenesis (1) High association with KRAS gene mutation (2) Mutation of suppressor genes (p16, SMAD4, p53, BRCA2) (3) d) Location: Most pancreatic head (65% of cases) e) Clinical & laboratory findings (1) Epigastric pain with weight loss (>90% of cases) (2) Blockage of common bile duct: jaundice, light color stool, palpable Gallbladder (3) Superficial migratory thrombophlebitis (Trousseau’s sign) (4) Metastasis to the left supraclavicular node (Virchow node), Late sign (5) Periumbilical metastasis (Sister Mary Joseph sign) (6) Marantic endocarditis (sterile cancerous nodules on cardiac valves) f) Laboratory findings: Increased CA19-9 tumor marker (1) Diagnosis: CT scan, Ultrasound guided endoscopic fine needle aspiration. (2) Gross/Microscopic: Fibrous mass obliterating pancreatic duct, Ill-formed glands with surrounding fibrotic stroma (3) Treatment: Surgery (Whipple procedure), radiation, chemotherapy g) E. Prognosis: Poor: 5yr survival rate is 20% [Some]Endocrine Pancreas 1. Islets of Langerhans, functional unit a) 1 million spread throughout pancreas b) Different cell types: (1) Alpha (α) cells producing glucagon (2) Beta (β) cells producing insulin & amylin (75%) (3) Delta (δ) cells producing somatostatin (4) Gamma cells (Pancreatic Polypeptide cells) regulatory (5) Epsilon cells producing ghrelin (hunger hormone) (<1%) 2. Diabetes Mellitus (See diabetes lecture for more detail on this topic) a) Lack of insulin production from pancreatic islets cell inhibition/destruction from various etiologies: metabolic, autoimmune, inherited syndromes, pancreatitis, neoplasms. b) Nonenzymatic glycosylation of host proteins alters function c) Widespread vascular damage: Kidney glomeruli, Cardiovascular, Eye, Peripheral nervous system, Liver, Skin d) 3. Microscopic: Islets - fibrosis hyalinization Islet cell tumors (Neuroendocrine tumors) a) Tumors seen with MEN-I (Multiple endocrine neoplasia, sipple syndrome) (1) Pancreatic islet tumors (2) Pituitary (3) Parathyroid hyperplasia b) Mostly benign, can be malignant (certain Ex-Apple exec) c) Can treat most with octreotide (somatostatin) at least until surgery. d) Insulinoma (β cell tumor) (1) Most common islet cell tumor (2) Marked increase in insulin secretion (3) Clinician distinguish endogenous insulin production from exogenous insulin administration (surreptitious or otherwise) via quantitate C-peptide (pro-insulin fragment) which is not present in exogenous insulin. (4) e) Clinical (Whipples triad) (a) Episodic hypoglycemia (b) CNS dysfunction (stupor confusion) from hypoglycemia (c) Reversal of CNS dysfunction by glucose administration Gastrinoma (1) Often malignant (up to 90%) (2) 2/3 are sporadic, 1/3 associated with MEN-I (3) Most common pancreatic tumor seen with MEN-I (4) Actually more common in duodenum than pancreas, also sometimes in meckels diverticulum (5) Gastrin hypersecretion & hypergastrinemia (Labs: Gastrin >1000 pg/ml) (6) Causes Zollinger-Ellison Syndrome (ZES) (a) Hypersecretion of HCl (b) Recurrent peptic ulcer disease: Ulcers in strange places is suspicious for ZES (c) Hypergastrinemia (d) f) Clinica: diarrhea, maldigestion of food Glucagonoma (α cell tumor) (1) Rare (2) (3) Clinical: Secondary diabetes mellitus (hyperglycemia) & skin lesion called Necrolytic migratory erythema g) Somatostatinoma (δ cell tumor) (1) Rare (2) Somatostatin inhibitory hormone, causes: (a) Achlorhydria (gastrin inhibition) (b) Stones & Steatorrhea (Cholecystokinin & secretin inhibition) h) VIPoma (Vasoactive intestinal peptide) (1) Very rare (2) Clinical: Pancreatic cholera: Watery Diarrhea, Hypokalemia & Achlorhydria [(WHDA) syndrome] (3) Labs: Hypokalemia, normal anion gap, metabolic acidosis (Bicarb lost in stool) Liver & Pancreas Case Studies Case 1 HISTORY: This 40-year-old woman developed severe, colicky right upper quadrant pain. For several years she had noted discomfort following ingestion of fatty meals. PHYSICAL FINDINGS: Obese woman in moderate distress; tenderness beneath the right costal margin. LABORATORY RESULTS: WBC: 13,000 /cu mm (nl: 4,000-11,000/cu mm) WBC diff: 81%seg, 1%bands, 1%Eos, 1%baso, 14%lymph, 2%mono Abdominal x-ray: distension of the gallbladder CLINICAL COURSE: She underwent an operation. The abdomen contained 200 cc of cloudy ascites. Postoperatively she complained of severe epigastric pain & was hypotensive. Next day LABORATORY RESULTS: Calcium: 6.5 mg/dl (normal 8.5-10 mg/dl); Amylase: 500 U/dl (normal 45-200); Lipase: 4.0 U (normal 0.5-1.5). She died on the third postoperative day. The original episodes of abdominal discomfort over the last few years were most likely due to what? The postoperative serum calcium value was most likely related to what? If the patient had survived, what would have been some of possible outcomes? Case 2 HISTORY: This 64-year-old man was admitted with a three week history of nausea, vomiting, food intolerance, dark urine, light colored stools, jaundice & 6 kg weight loss. Abdominal pain was minimal to absent. PHYSICAL FINDINGS: liver mildly enlarged to percussion, fluctuant abdominal mass in right upper quadrant of abdomen thought to be distended gall bladder, spleen not palpable. LABORATORY FINDINGS: Alkaline phosphatase: 400 U/L (normal 30-115) Bilirubin, total/direct: 12.9/10.9 mg/dl SGOT (AST): 53 mU/ml (normal 8-40) Serum protein, total/albumin: 6.8/3.7 g/dl What would expect to be the most likely finding on liver biopsy? At surgery, the finding that would fit best with the clinical profile would be what? Would you expect the patient’s urine to be positive for: urobilinogen (?), conjugated bilirubin (?), unconjugated bilirubin (?). The most likely diagnosis in this case is? Case 3 This 52-year-old lawyer had malaise & anorexia & fatty food intolerance for 2 weeks before admission, dark urine & vomiting for 1 week, jaundice & light colored stools for 3 days. He had lost 12 kg in the previous six months, drank 15-20 cans of beer per week, & had been exposed to a person with jaundice one month previously. LABORATORY RESULTS: Hemoglobin-16.0 g/dl bilirubin-5.2/8.1 mg/dl SGOT (AST)-250 mU/ml Serum albumin-3.4 g/dl Serum gamma globulin-3.3 g/dl; Alkaline phosphatase-180 mU/ml (normal 30-85). The disease process which best characterizes this clinical scenario is? Case 4 HISTORY: This 20-year-old student was admitted with a 10-day history of chills, fever, nausea, vomiting, dark urine, fatigue, & a 2-day history of jaundice. He had been living in a communal setting for the last 10 months & admitted using marijuana & LSD. PHYSICAL FINDINGS: Tenderness in the right upper quadrant, excoriations of the pubic area & scrotum. LABORATORY RESULTS: Hemoglobin: 14.2 g/dl WBC: 5,400 /cu mm Bilirubin, total/direct: 12.0/6.2 mg/dl Alkaline phosphatase: 250 U/L (normal 30-115) SGOT (AST): 1120 mU/ml (nl:8-40 mU/ml) LDH: 240 mU/ml (nl:110-212mU/ml) CLINICAL COURSE: The patient refused hospitalization, & disappeared after percutaneous liver biopsy was performed. What other laboratory tests would help you confirm a diagnosis in this case? The cause of this patient's illness is most likely due to what? Urine would be expected to be positive for: bilirubin (?), bilirubin (?) Case 5 This 17-year-old girl had an episode of jaundice & malaise at age 8. One year before admission she developed jaundice, fatigue, acholic stools & dark urine which lasted for six weeks. Four weeks before admission she again developed jaundice & one week later vomited bright red blood. Physical exam: revealed a slightly enlarged liver & a very large spleen. Imaging: Esophageal varices were seen on barium swallow. LABORATORY RESULTS: Hemoglobin-10.2 g/dl WBC-5,300/mm3 bilirubin-1.1/1.7 mg/dl Alkaline phosphatase-320 mU/ml (normal 30-85) SGOT (AST)-460 mU/ml; SGPT (ALT)-126 mU/ml. A splenectomy & porto-caval shunt was done. The disease process which best characterizes by this clinical scenario is? What do you suspect is the etiology of her current illness? What other laboratory test or procedure would help you confirm the diagnosis? Case 6 This 58-year-old man had intermittent thrombophlebitis in both legs for two years. Two weeks before admission scleral icterus was noted along with the vague feeling of weakness. Physical exam revealed a fluctuant distinct mass just below the right rib margin, but no other abdominal findings. LABORATORY RESULTS: Bilirubin-12.0/15.7 mg/dl Alkaline phosphatase-500 mU/ml SGOT (AST)-55 mU/ml Albumin-4.0 g/dl Globulin-2.8 g/dl. An exploratory operation revealed an irregular firm mass compressing the lower common bile duct. 1. Diagnosis? 2. What else is needed to confirm? 3. Outcomes? Case 7 HISTORY: This 53-year-old woman was admitted with anorexia & nausea for one week & jaundice for 2 days. She had felt well since having a mitral commissurotomy for rheumatic valvulitis 5 months ago. She had stopped smoking & discontinued her daily cocktail since onset of the anorexia. PHYSICAL FINDINGS: Normal except for sternal scar & jaundice. LABORATORY RESULTS: Hematocrit: 42% WBC: 9,500 /cu mm Urine bilirubin: positive Urine urobilinogen: positive Bilirubin, total/direct: 9.4/8.8 mg/dl SGOT (AST): 920 mU/ml (normal 8-40) Alkaline phosphatase: 110 mU/ml (normal 30-115) Protein, total/albumin: 6.8/3.9 g/dl prothrombin time: 15 sec (normal 11-13) 1. The most likely diagnosis? 2. What’s needed to confirm? 3. Long-term complications in patients with this disease include? Case 8 HISTORY: This previously well 42-year-old woman presented with a 2-day history of crampy right upper quadrant abdominal pain. She had been using oral contraceptives for the past 4 years. PHYSICAL FINDINGS: Mildly obese woman in moderate distress. The abdomen was tender in the right upper quadrant. Liver & spleen were not palpable. LABORATORY RESULTS: Fasting glucose: 115 mg/dl Bilirubin, total/direct: 2.0/1.6 mg/dl Urine bilirubin: positive Urine urobilinogen: negative CLINICAL COURSE: She was treated with nasogastric suctioning & intravenous fluids with good response. A repeat bilirubin the next day was 2.4 mg/dl. 1. The most likely diagnosis? 2. The urinary findings would be compatible with? Case 9 This 62-year-old man was admitted 6 days before death in a semi-comatose state & no history was obtainable. He was severely jaundiced & had a positive abdominal fluid wave. LABORATORY RESULTS: Bilirubin-18.6/22 mg/dl Alkaline phosphatase- 200 mU/ml (normal 30-85) SGOT (AST) 435 mU/ml. His condition worsened & 2 days before death he developed a fever & leukocytosis. 1. The disease process which best characterizes these pictures is? Case 10 HISTORY: This 36-year-old male automobile salesperson was admitted to the hospital complaining of hematemesis, melena & extreme weakness of 3-months duration. He had sustained a laceration of the liver capsule in an automobile accident ten years earlier, at which time his liver was described as enlarged soft & abnormally light in color. He was hospitalized four years earlier for an episode of jaundice, during which a hepatic needle biopsy revealed well-advanced, micronodular cirrhosis. He admits to having been a "moderate-to-heavy" drinker but has abstained from alcoholic beverages since his last hospitalization. PHYSICAL FINDINGS: Pale skin; slightly icteric sclerae; liver not palpable but spleen enlarged & firm; gynecomastia; testicular atrophy. IMAGING RESULTS: Barium swallow radiography-evidence of esophageal varices CLINICAL COURSE: Despite attempts to control hemorrhage the patient developed irreversible shock & died on the fifth hospital day. Autopsy revealed 1,200 ml of clear straw-colored fluid in the peritoneal cavity. 1. Microscopic examination of the liver biopsy specimen at the time of his automobile accident would most likely reveal an excess of? 2. The feature ultimately most responsible for the fatal upper gastrointestinal hemorrhage is?