Supplementary Information
advertisement

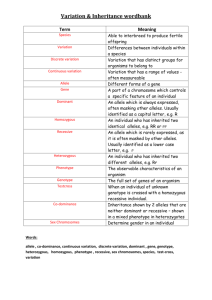
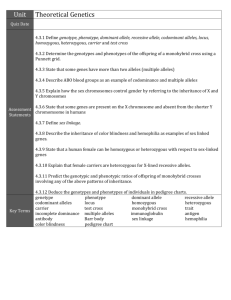
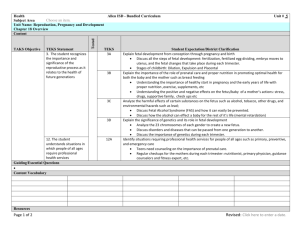
Supplementary Information Model We are interested in modeling the statistical limitations of cell-free DNA based diagnostics for Mendelian and mainly recessive diseases. Since the amount of fetal DNA in maternal blood varies widely and increases over the course of pregnancy, it is uncertain whether most early gestation cases will have sufficient fetal DNA to derive a statistically meaningful result. In selecting the correct model, we think of the different DNA alleles of a target region the same as different-colored discrete marble balls in an effectively infinitely large bag. The bag, representing the cell-free portion of the circulatory system, would be well mixed and have many copies of both marbles or alleles (as an estimate, one target would specifically have about 1000 copies/mL * 5000 mL plasma = 5 million copies in the entire plasma circulation). A standard blood draw with a subsequent unbiased counting measurement would be similar to blindly pouring out of the bag and thereafter counting up the different colored marbles. We assume that outflow of a particular allele or marble is not going to affect the probability of what other marbles will be. Then we can assume that the outflow of the 2 independent marbles/alleles follows a Poisson distribution and are independent of each other. To aim at the theoretical maximum, we assume perfect technical execution with no bias in the process to either allele. We also assume that the mother is a carrier and is therefore heterozygous at the mutation site. The calculations based on Poisson distributions of finite DNA allele counts of a target allele are below: Definitions: fetal _ DNA total _ DNA NT = # total alleles = NM + NW, NM = # mutant alleles, NW = # wildtype alleles 1 Figure S1: Cell-free DNA can be divided into DNA from a fetal cell origin or maternal cell origin. The fraction that is fetal cell derived is fetal fraction (diagonal lines). In any scenario, the mother contributes equal amounts of both alleles. A fetus that is heterozygous or a silent carrier (and unaffected) also contributes equal amounts of DNA. However a fetus that is homozygous contributes all its DNA or ε * NT additional DNA to the homozygous allele count. Based on Figure S2, we can infer that: If fetus is homozygous case then on average: NM - NW = ε * NT If fetus is heterozygous case then on average: NM - NW = 0 We then calculate the standard deviation for each allelic count in both scenarios and then calculate the combined standard deviation after the subtraction NM - NW (the variance of sum of two normal distributions is equal to the sum of the two variances). We find conveniently that in both scenarios the standard deviation is the same at NT . Homozygous case: σ of NM = 0.5 * NT 0.5 * σ of NW = 0.5 * N T 0.5 * σ of NM - NW = = ( 0.5 * N T 0.5 * ) 2 ( 0.5 * N T 0.5 * ) 2 NT Heterozygous case: 2 σ of NM or NW = 0.5 * N T σ of NM - NW = ( 0.5 * N T ) 2 ( 0.5 * N T ) 2 = N T (same as the homozygous case) The Z-statistic or Z-score can be calculated by putting the allelic count difference over the common standard deviation. Given that the denominator is the same, the Zscores can be compared regardless of fetal genotype. With the below simplified analytical equation we can generate curves relating Z-score to fetal fraction (ε), and total counts (Figure S2). Z score (theoretical) = * NT NT Equation S1: Theoretical average Z-score based on a Poisson approximation for a single SNP when the fetus is homozygous. Z score (empirical) = Equation S2: NM - NW NT Empirically derived Z-score for a single SNP when the fetus is homozygous. The allele counts that we use for Equation S2 refer to the lowest amount of molecular counts in the entire sample processing and method. For example if a single target was counted one million times after an amplification step but it only had 1000 molecules prior to amplification then the total counts should be renormalized to 1000 rather than 1 million and all calculations should be based values recalculated on basis of the 1000 value. The calculations here are for genetic content that is measurable. An example of genetic content that is in the plasma but not measurable is an allele physically located at the edge of a DNA fragment and therefore would not be amendable to PCR amplification (although measurable with sequencing). An 80 bp amplicon will only amplify about half 3 of the strands contain in the targeted allele given that the allele location is evenly distributed on the stereotypical 160 bp strand. Published efforts have shown that shorter amplicons can effectively enrich for fetal content presumably because fetal DNA fragments are shorter1. One key point here is that some samples may lack the fetal fraction and blood quantity to reach the minimal theoretical threshold for confidence. Other samples may reach the minimal theoretical threshold but they will have overlap between the theoretical distribution of a homozygous and heterozygous fetus. A measurement that falls by chance into the overlap would be indeterminate. There is a range of 1000-2000 copies per mL plasma (~500-1000 copies per mL blood) and when fetal fraction 5% (average value for first trimester), one needs a 20,000 copies or 20 mL of blood to achieve good separation of the fetus homozygous and fetus heterozygous scenarios. At 2%, it is likely that more than 100,000 copies (~100 mL) will be necessary to ensure that more than 99% of samples will be distinguishable—a requirement that is unlikely to be practical. In routine practice, a tube may contain 10 mL of blood, and although several tubes are routinely drawn for pregnant women, many of them are used for a battery of other routine tests. While a blood donation is much more volume at 1 pint or about 450-500 mL, the logistics of transportation and sample processing are practical barriers. Luckily for the vast majority of cases, it may be that only a few tubes of blood are necessary and this or a related model will be employed to ensure that the result is statistically confident for one fetal genotype and does not fall into a region of overlap between the distributions of two fetal possibilities. While we have described the case of a homozygous mutation, recessive Mendelian diseases can occur as a compound heterozygous combination of 2 alleles on different locations on the same gene. For example, one mutation can be a premature stop codon on exon 2 and another could be a point mutation in exon 5 in a critical active site of the gene’s protein. Compound heterozygous states will effectively disable both copies of the same critical gene and can occur frequently depending on the population in question. The same essential model and equations presented above can be used for compound heterozygous scenarios. For these scenarios, it is critical to distinguish between heterozygous and homozygous non-diseased allele at maternal mutation site. If 4 the maternal site is heterozygous and the paternal site also has a mutation, then it would imply a disease phenotype. Measuring the paternal site is less technically challenging and similar to the measurement of the fetal fraction. The model described can also be applying to multiple haplogroup linked markers. If the markers are assumed to be 100% associated with the mutation then the allelic counts of each marker are summative. Figure S2: Theoretical Z-score averages for various molecular counts and fetal fractions if the fetus is homozygous (based on Equation S1). Heterozygous Z-scores always average zero. Confident calls involve either high Z-scores from a homozygous fetus or near-zero Z-score from a heterozygous fetus. The Z-score distribution of the two fetal genotypes will not significantly overlap when an average theoretical Z-score of a homozygous fetus is over 4 (almost all of each distribution is within 2 Z-scores in each 5 direction). There with a fetal fraction of 15%, 1400 counts would result in a confident call in almost all cases; for fetal fraction of 5%, 20,000 counts would be necessary. However, even if with some overlap between the two distributions, the empirically counted Z-score can still fall outside the zone of overlap by chance and result in a confident diagnosis. To take into account haplotype-linked SNPs, the counts of alleles from other loci linked to the mutation are summative. For example, 1000 counts from 10 sites will be effectively 10,000 counts. Note that this assumes negligible amplification bias when amplifying the haplotype-linked loci. Figure S3: Example readout of droplets from a cell-free sample with two alleles that correspond to the two respective fluorophores FAM and VIC. 6 Table S1: Markers for mutation and haplotype linked positions, their digital PCR counts, and calculated Z-scrore. Probe # dbSNP # Wildtype Allele Wildtype Counts Mutation Counts Total Droplets G Mutation associated allele A 249 (direct) rs1219182 57 656.2 927.4 43939 249 (postPCR) 213 214 218 219a 219b 223 rs1219182 57 G A 5440.0 3807.6 25520 rs4715130 rs6923124 rs2229384 rs7750918 rs3729619 rs4469291 C G T TA[G]TT CG[A]AG AATTTTT[A]A A G G T T 233 234 241 243 rs7774688 rs4573082 rs497734 rs9369836 Normaliz ed Zscore 5.97 7.12 T A C TA[C]TT CG[T]AG AATTTTT[T]A A A T G C 2099.6 3521.0 1288.0 3473.0 1774.0 2532.9 1665.9 4555.4 1534.3 4565.7 2031.6 3223.3 27798 27897 25520 27262 28103 27557 4.56 5.58 3.28 4.88 3.82 4.46 2556.8 1954.2 3010.7 2768.2 3208.0 2337.6 4024.9 3467.7 27847 27798 27897 28427 3.28 2.80 4.72 5.15 1. Sikora A, Zimmermann BG, Rusterholz C, Birri D, Kolla V, Lapaire O, Hoesli I, Kiefer V, Jackson L, Hahn S: Detection of Increased Amounts of Cell-Free Fetal DNA with Short PCR Amplicons, Clinical Chemistry 2010, 56:136-138 7