Pi bonding 2
advertisement

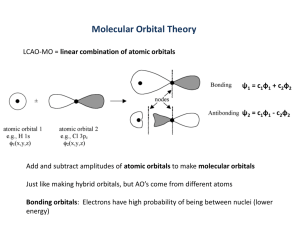
bonding in linear alkenes We will start with two C atoms and work our way up: Consider H2CCH2 a 2 carbon chain. We know that already it is H2C=CH2. We note that the Eneregy C's are sp2 hybridized. There is one bond and it 2px is not aromatic. - After we use the sp2 orbitals to form the sigma frame work we have one p orbital on each carbond left over for the orbitals: 2px 2px However if we look at it from a bonding point of view we have two possibilities: The last p orbital on each C are show below. + 2px + + C C - - Now we can take positive and negative contributions to make our wave functions and orbitals. Take a empty circle as a posit ive lobe of px orbit al coming out of paper positive contribution and a filled circle as a negative contribution. we can schematically show the negat ive lobe of px orbit al coming out of paper orbitals as: C C Nodes 0 1 The top line corresponds to taking a linear combination of the two p as shown below on the left while the second line corresponds to the linear combination on the right: By the way the MOs look like: Or to make it simpler use + and – signs for the contribution of the AO's C + + Nodes C + - 0 1 We have written it with the number of nodes going down so remember that if you want to write the orbitals in terms of energy you need to write it like: C + + Nodes C + - + 1 0 Where is the energy of the AOs and is the amount that the bond stabilizes or destabilizes the MO. Note that we can not really do three carbons because H2CCHCH2 is a radical and the last C does not have a full octet. Draw the Lewis dot structure and you will see. It will react to form H2CCHCH3 and now the last carbon is sp3 and not sp2. Now we go on to four carbons H6C4 where again all C's are sp2 hybridized: H H C C H H C C H H C + + + + C + + C + + C + + Nodes 3 2 1 0 + 1 .6 + 0 .6 - 0 .6 - 1 .6 You note that if you draw the Lewis dot structure you get two double bonds and one single bond between the Cs. In the MO bonding scheme above you have 4 electrons (one from each C). With 4 electrons you fill up the two lowest orbitals. Both these levels have a bond between the two C atoms on the left and the two on the right. The lowest level also has a bond between the two central C atoms but the next orbital is antibonding for the two central atoms. Thus this MO (remember this is only the orbitals we are talking about and the orbitals already have a bond between each C atom) has bonds between the end C atoms and not much extra between the two central C atoms. Thus the lowest orbital contributes bonding between each of the carbon atoms. The second orbital has bonding between the first and second C and a nodebetween the second and third. The net result is that for 2 MO’s we get extra bonding between the end C’s and some extra between the center two. In the molecule the bond length between the two end carbons is close to that of a double bond but a little less and the center two carbons distance is a little shorter that a single bond. Look at the pluses and minuses in the table above as a matrix. You see that: The first column and the bottom row are the same. So is the second column and the third row, etc. It is symmetric along a line from the low left to the upper right. So what are the rules for drawing the MO picture of the bonds: A) Keep the rows with odd number of nodes symmetric to inversion (+ - | - +)) B) Keep rows with even number of nodes antisymmetric to inversion (+ - |+- -) C) Column i is the same as the reverse of the row with nodes i-1 (first column (all +) is same as bottom row (all +) , second column (- - + +) is same as second row(+ + - -) Procedure: 1. Start at the bottom and work up. 2. Write the zero node row as all + on the bottom. 3. Write the first column as all + 4. Write the node 1 row above the node zero row as half + and half – 5. Copy this to the second column 6. Use first part of row with 2 nodes to fill in end of row (remember it is symmetric) 7. Fill in rest of row with 2 nodes to make 2 nodes 8. etc Ok now try do an 8 carbon chain: C + + + + + + + + C + + + + C + + + + C + + + + C + + + + C + + + C + + + + C + + + + Nodes MO’s 7 6 5 4 3 2 1 0 There is another way to write it also that follows the rules: - 1 .9 - 1 .5 - 1 .0 - 0 .3 0 .3 6 + 1 .0 + 1 .5 + 1 .9 C + + + + + + + + C + + + + C + + + + + + C + + + + C + + + + C + + + + C + + + + C + + + + Nodes 7 6 5 4 3 2 1 0 And a number of others! At this level it does not matters since you cannot tell them apart. The two ways of writing the 2 node orbitals actually are not exactly correct and a linear combination of the two ways is needed but don't worry about that. Energies of the levels for the linear Hn+2Cn hydrocarbons: are listed below for information note that all levels are not degenerate: n 2 3 4 5 8 10 20 lowest level center Highest level Note that as n gets bigger the lowest and highest orbitals are closer and closer to and that the spacing between orbitals becomes smaller. This is the beginning of the band structure that is seen in semiconductors and metals. I have put in the odd numbered length molecules but these are not stable to reacting and adding another CH bond. However you can talk about the radical and so they have been added. You note that for even length C compounds you do not get any orbital that is in the center and that the orbitals have an even spacing in energy. bonding in Cyclic Alkenes Now we will try cycles, the nodes are shown in red below here are the rules: 1. Draw the outline of the compound with one atom down. 2. Put a line at the level of every vertex or C atom in the compound, these are the MOs. 3. Make a table showing the contributions to each MO as above. 4. The orbitals lower than the center are bonding orbitals and the ones higher are antibonding orbitals Remember that for an odd number the molecule is a radical (i.e. one C does not have a full octet). Here is H6C3 C — C — C nodes + - + 3 1 + + + 1 - 1 + 0 Energy t ype - - + 2 2 Now remember that H3C3 is a radical and would like to form H4C4. But the radical has three electrons and the last electron is in an antibonding orbital. Thus it only has about a half of a bond and that is not great. Also since the highest two orbitals are degenerate (they have the same energy) the molecule would like to distort. It will distort since the top 2 orbitals are degenerate if it distorts that will remove the degeneracy and so one orbital will become more stable and one less stable. Since you have only one electron in the top orbital that electron will go into the more stable lower orbital and make the molecule more stable. The distortion will involve one of the C-C bonds becoming a double bond and the other two being single bonds. This sort of distortion is important later. Here is H8C4 C — C — C —C nodes Energy t ype + + + + + - + - 2 1 1 + + + + 0 - 1 2 + 2 1 This is a true molecule and not a radical. It has a bonding and an antibonding orbital and two nonbonding orbitals. There are two electrons in the bonding orbital and two in the nonbonding orbitals. Thus it really has only one extra bond and so it is not any better (actually worse) then the Lewis structure that has two double bonds and two single bonds. Again since the two nonbonding orbitals are degenerate they can distort and since there are two electrons they will go into the orbital that become more stable H H and pair. Thus this molecule will distort, it will not be a square more C C likely a rectangule with have two long C-C bond and two short C-C bonds, seee figure at right. The bonds become localized and are not spread out over the whole molecule. It is also then not aromatic. C H5C5. C H H C — C — C — C — C nodes + - + 1 + - + + + + + - + 2 - + - 2 - - + 1 - - 1 + + 0 + 1 .6 1 .6 +0 .6 +0 .6 + 2 Here again we have a radical when it is netural. The bonding has three bonding orbitals and two antibonding. For the neutral we have five electrons in the bonding orbitals so that is pretty good, however the partially filled bonding orbitals are degenerate and so the molecule will probably distort to split that degeneracy and put two electrons in the lower and one in the higher. It will also react to form H6C5, which is not a radical and is shown below. H H H H C C C C C H The anion, H5C5–, has an extra electron and now there are 6 bonding electrons and so it is stable, planar and it is aromatic. If we consider the cation H5C5+, the position ion we now have 4 electrons in the degenerate bonding pair and so it is again not stable. It will distort to form two double bonds and three single bonds. H 6 5 1 2 4 C— + + + + + + 3 C— + + + + + + + C— + + + + + - C— C— + + + + + + C— + + + + C— + + + + - C— + + + - C — C nodes + + 0 1 + 1 2 + 2 + 3 C— + + + C— + + + + - C— + + + + + C nodes + 0 1 + 1 2 2 3 3 4 1 2 Here we have benzene, H6C6. It has 6 p orbitals and 6 electrons and they fill up the three bonding orbitals. Thus it is planar and aromatic. The H8C8, has 8 p orbitals and 8 electrons. The fill up the three lowest level and the last two electrons go into the nonbonding degenerate pair. Again we have a situation that there is an unfilled degenerate orbital and it will want to distort. The molecule will distort to have four double bonds and four single bonds, shown to the right. Energy levels for cycle hydrocarbons HnCn note that most levels other than the first and last are doubly degenerate. The last level for n odd is also degenerate n lowest Middle Highest level Level level 3 4 5 6 7 10 20 … Note that the lowest orbital is always at which is the limit for n large in straight chain HC's. If we look at a cyclic hydrocarbon as just a straight chain hydrocarbon that has a bond from the last C to the first (physicists call this cyclic boundary conditions) then we see that once you do this the lowest energy orbital is at the energy for a straight chain hydrocarbon that has a very long chain. All the odd numbered carbon cyclics have a double degenerate highest orbital and no orbital that is nonbonding All the even number orbitals that have 4n carbons (4,8,12,16,…) have a nonbonding orbital that is partially filled. This means that they are not aromatic. All the even number orbitals that have 4n+2 carbons (6,10,14,..) have no nonbonding orbirtal and have half of their orbitals as bonding and half as nonbonding. Since they have the same number of electrons as orbitals half of the orbitals are filled and half are empty (remember that orbitals take 2 electrons). Thus these cyclic compounds have filled boning orbitals and like to be planar and are aromatic.