REDUCTION OF ALDEHYDES, KETONES,TO
advertisement

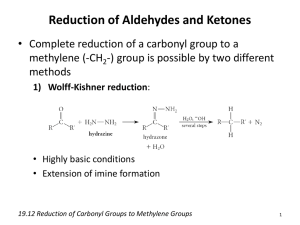
REDUCTION OF ALDEHYDES, KETONES,TO Aldehydes and ketones can be reduced to hydrocarbons, alcohols, and pinacols [1, 2 diols or glycols]. [A] HYDROCARBONS Five methods are available. The choice between them is based on the sensitivity of other functional centers in the reactant in the reducing conditions. [1]Clemmsens Method The reducing agent is amalgamated zinc and concentrated hydrochloric acid. The probable mechanism is as follows: The concentrated acid is apparently needed to force the initial protonation: amalgamation of zinc raises the hydrogen-overvoltage so that hydrogen is not produced. Only halogen acids are effective, probably because, by complexing the initial –Zn+ specie, they provide a medium for the reduction of this species by the second atom of zinc. The method is particularly useful for ketones which contain phenolic or carboxylic groups. For example, the reduction of β-benzoylpropionic acid in toluene gives γ-phenylbutyric acid in 85% yield This type of reduction forms one step in the extension of benzenoid systems via Friedel-Crafts acylations. The reagent also reduces the olefinic bond in α β–unsaturated ketones, acids, and esters; and benzyl halides and alcohols are hydrogenolyzed. Strongly hindered ketones give low yields and sometimes rearrangement products [e.g. Ph3C-CO-PhPh2C=CPh2] [2]Wolff-Kishner Method. The hydrazones of aldehydes and ketones are reduced in vigorous basic conditions with the evolution of nitrogen, probably as follows: The standard procedure is the Huang-Minlon modification. The hydrazone is formed by heating the carbonyl compound with hydrazine hydrate and potassium hydroxide in di- or tri-ethylene glycol under a water condenser. After completion of the formation of the hydrazone, the water condenser is removed so that water liberated in the first reaction is distilled and the temperature rises to about 200 oC, so bringing about decomposition of the hydrazone. A newly developed modification employs potassium t-butoxide as the base and dimethyl sulphoxide as solvent. Alkoxide bases are very much more powerful in this solvent than in water or hydroxylic solvents and reaction occurs at room temperature in high yield. For example, benzophenone gives about 905 of diphenylmethane: [3]Mozingo Method. The carbonyl compounds are converted with ethylene dithiol in the presence of a Lewis acid into its dithiol-acetal or ketal and this is hydrogenolyzed over Raney nickel: Alternatively, the cyclic ditio compound is reduced by hydrogen-transfer from hydrazine at 100-200C. The Mozingo reaction is useful for reducing carbonyl compounds which are sensitive to mineral acid and bases, for the Clemmensen and Wolff-Kishner methods are then unsuitable [4]Tosylhydrazone Method. Reaction of carbonyl compound with toluene –p-sulphonylhydrazine gives the tosylhydrazone which is efficiently reduced by sodium borohydride [Ar=ptolyl]: For example, the keto group in androstan-17β-ol-3-one is reduced in this way in about 75% yield Reaction probably occurs as follows: [5]Lithium aluminum hydride Aromatic ketones are reduced by lithium aluminium trichloride. Reaction occurs by reduction to the alcohol followed by hydrogenolysis of the benzylic system, aided by Lewis acid: [B]TO ALCOHOLS Carbonyl compounds are reduced to alcohols by the variety of reagents. Of the three general classes of reductive process, catalytic hydrogenation is not normally chosen because it is slow, but both hydride-transfer reagents are employed. [I]HYDRIDE TRANSFER The alkali-metal hydrides such as sodium hydride are unsuitable reducing agents because of their insolubility in organic solvents and their powerful effects as catalyst for base-catalyzed condensations. The most commonly used hydride reducing agents are lithium aluminium hydride, sodium [or potassium] borohydride and lithium borohydride. [1Lithium aluminium hydride Lithium aluminium hydride is made by treating lithium hydride with aluminium trichloride in ether, and is generally used in very dry ether or tetra-hydrofuran .All hydroxyl-,amino-,and thiol-containing compounds liberate hydrogen quantitatively from it e.g., Each of the four hydrogen atoms in lithium aluminium hydride is available for the transfer to carbonyl groups. e.g. each step occurring less rapidly than the proceeding one[As result, the replacement of two or three hydrogen atoms of aluminium hydride anion by alkoxy groups give less reactive selective reducing agents;].Finally, hydrolysis of the aluminum alkoxide give the alcohol. [2]Sodium borohydride. Sodium borohydride is much less reactive .It can be used in alcoholic solvents and even in water ,for it decomposes only enough to make the solution alkaline, after which it is stable. [3]Lithium borhydride Lithium borohydride in more reactive than sodium analogue and reacts with hydroxylic compounds. It is usually employed in solution in tetrahydrofuran or diethylene glycol dimethyl ether [diglyme]. These three reagents differ considerable in their reducing power. Lithium aluminium hydride reduces not only aldehydes and ketone but also acids, acid chlorides, esters, nitriles, imines, and nitro groups, whereas sodium borohydride reduces only aldehydes, ketones, imines and acid chlorides. Lithium borohydride resemble sodium borohydride except that it also reduces esters and nitriles. None of the reagents normally reduces olefinic, acetylenic, or N=N bonds, although the first reduces acetylenes containing αhydroxy-substituents and reduces azo compounds in the presence of Lewis acid. Except in simple cases, therefore, sodium borohydride is the reagent of choice. Typical examples are: In rigid ring systems, the stereochemistry of hydride reduction appears to be determined usually be the relative importance of competing influence; steric hindrance to the approach of the reagent, and the stability of the final product. Unless steric effects are particularly severe, the latter factor dominates; for example, 10 –methyl-2-decalone-gives mainly the more stable equatorial alcohol, although this involves approach of the reagent from the more hindered side; [4]Cannizarro reaction. Aldehydes which do not have α-CH groups cannot undergo base-catalyzed condensation. Instead they react with bases by disproportionation involving the transfer of hydride ion e.g. Crossed Cannizarro reaction between one such aldehyde and formaldehyde result in the reduction of the former and the oxidation of the latter, for formaldehyde is more reactive than other aldehyde toward nucleophiles and rapidly gives a high concentration of the donor anion: This fact can be exploited for reduction .For example, benzaldehyde is reduced to formaldehyde in the presence of potash in refluxing methanol to give 80% of benzyl alcohol. The preparation of pentaerthritol from acetaldehyde and formaldehyde is also dependant on a crossed Cannizarro reaction. [5]Meerwein-Ponndorf-Verley reaction This reaction is the reverse of the Oppenauer oxidation: equilibrium is established between the carbonyl group to be reduced and isopropanol on the one hand, and the required alcohol and acetone on the other hand, in the presence of aluminium iso-peroxide .Since acetone is the lowest boiling constituent of the mixture, it can be continuously distilled so that the equilibrium is displaced to the right. For example, trichloroacetaldehyde to trichloroethanol in about 80% yield. The reaction is specific to aldehydes and ketones; in particular, the olefinic double bond is αβ-unsaturated aldehydes or ketones is not reduced [compare lithium aluminium hydride. However, the basic conditions may bring about side-reaction [as in the synthesis of reserpine]. Very hindered Grignards reagent effect reduction in similar way. [II]ELECTRON TRANSFER REAGENTS These reagents are less selective than sodium borohydride and the Meerwein-Ponndorf-Verley reagent; e.g., they also reduce the olefinic double bond in αβ-unsaturated carbonyl compounds. Nevertheless, in simple cases they are rapid and efficient; e.g., methyl n-amyl ketone is reduced by sodium in ethanol to 2-heptanol in over 60% yield, and n-heptaldehyde is reduced by iron in aqueous acetic acid to n-heptanol in 80% yield. These reductions are stereoselective. In most cases, the thermodynamically more stable alcohol predominates: for example , 2methylcyclohexanone gives mainly trans-2-methyl-cyclohexanol. The reason is not fully understood, but one possibility is as follows: the metal transfers one electron to the carbonyl group to form an anion-radical, this is protonated at carbon fro the less hindered side, and the second electron is transferred to give the [usually less stable] alkoxide ion. This reacts with more of the ketone by a mechanism similar to that in the Meerwein-Ponndorf-Verley reduction above, giving an equilibrium mixture favourable to the more stable alkoxide, so that the final hydrolysis gives the more stable alcohol. However, if the ketone is very reactive as a result of stain ,the rate of the direct reduction may be so much greater than that of the attainment of the final equilibrium that the less stable alcohol is formed predominantly ,as in the reduction of camphor: If the ketone contains at the α-position a substituent which is good leaving group, the intermediate anion undergoes elimination: Zinc is often used as the electron source in these reductions. For example, αhydroxycyclodecanone gives cyclodecanone in about 75% yield when treated with zinc in mixture of hydrochloric and acetic acid at 75-80 oC [C] PINACOLS. In the absence of a proton-donor, electropositive metal reduce ketones to pinacols via the dimerization of anion-radicals: The standard procedure employs amalgamated magnesium with benzene a solvent; the solid magnesium salt of the pinacol is formed and is hydrolyzed to the pinacol. For example, acetone gives pinacol itself in 45% yield after 2 hours in refluxing benzene The reaction is generally ineffective for aldehydes because they are too readily reduced to alcohols. Pinacols may also be formed by photochemical dimerization [D] REDUCTION OF EPOXIDES, [a]Lithium aluminum hydride Epoxides are reduced to alcohols by lithium aluminium hydride. Since epoxides are readily obtained from olefins , the overall reaction serves to hydrate the olefin. The procedure is complementary to the hydroboration method since the hydride selectively attacks the less alkylated carbon of the epoxide ring, so giving the more alkylated alcohols The reaction has the trans stereochemistry characteristics of S N2 reaction. Thus, in a rigid cyclic systems the axial alcohol is formed, e.g., This therefore complements the methods for obtaining the equatorial alcohol by reduction of the corresponding ketone with electron-transfer reagents. The stained ring in four-membered cyclic ether is also cleaved by lithium aluminium hydride e.g., But the near-stainless five-membered ethers are resistant. They are however, opened in the more vigorous conditions obtained by using lithium aluminium hydride in the presence of the aluminium trichloride e.g., tetrahydrofuran gives n-butanol: [b]Hydroboration Epoxide are reduced by diborane to give mainly the less substituted alcohols e.g., The method is therefore complementary to the use of lithium aluminium hydride. With 1-alkylcycloalkane epoxide, the main product is the cisdisubstituted alcohol, e.g., This therefore complements the reaction of the 1-alkycyclohexames with diborane followed by alkaline hydrogen peroxide, which yields the trans product.