IP Buffer
advertisement

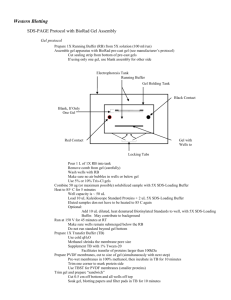
IP Buffer To PBS add, 10mM EDTA .1%Triton-X 100 1mM PMSF (To make 50mls, add 1ml of .5mM stock EDTA, 0.5ml of 10% stock of Triton-X, and .5ml of 100mM stock of PMSF) Thaw PMSF before using 1X-TBST 20mM Tris 150mM NaCl .1% Tween 20, P.H 7.4 Blocking Buffer 5% BSA in TBST 50mls TBST:2.5g BSA I. Biotinylation 1. Wash cells with PBS at room temp 3x’s 2. Incubate cells with 0.5mg/ml of sulfo-NHS biotin in PBS at room temp for 30 minutes (put 1ml on each gel and incubate on rocker) 3. Wash cells with PBS at room temp 3x’s II. Extraction/Lysis 1. Put 500ul of IP Buffer(lysis buffer) into 1.5ml centrifuge tube 2. Scrape gels with cells into the 1.5ml centrifuge tube (keep on ice) 3. Vortex and incubate on ice for 10 minutes 4. Centrifuge for 5 min at 4 degrees and 14k RPM 5. Keep lysate (liquid) 6. May need to resuspend in 500ul, and spin down again to get all the lysate (liquid) 7. Spin down the centrifuge tube containing all the lysate to make sure there is no acrylamide in the solution 8. Divide up lysate III. Add Antibody 1. Add 10ul of antibody to lysate and let incubate overnight at 4 degrees( If not adding antibody to the lysate just keep in IP buffer for overnight) IV. Add Beads 1. Add 75ul of Protein G Sepharose to the suspension that contains antibodies 2. If you have lysate without antibody add 75ul of Strepavidin (make sure beads are mixed 3. Incubate in beads for 1 hr at 4 degrees 4. Spin down solution, remove sup (and save) and wash beads 2x with IP buffer 5. After washing beads, add 50ul of 2xsds page sample buffer to each bead 6. Boil for 5 minutes to release the proteins from the beads 7. Spin down 8. Remove the sup from the beads *****this is what you want to run in the gel V. Run Gel 1. Remove comb from the gel and wash out wells 2. Dry and mark the bottom of the wells 3. Remove the paper tab 4. Insert with the gels with the ridge facing the center of the apparatus, and lock down 5. Fill inner chamber with SDS running buffer 6. Fill ½ of the outer chamber with SDS running buffer 7. Put 20ul of marker and protein in the wells 8. Turn the current all the way up, and set the voltage to 100 v—after it gets through the stacking gel, set it to 150 v VI. Transfer 1. Crack open the gel apparatus 2. Let gel stick to one side 3. Remove the stacker gel 4. Put gel in transfer buffer by using gravity 5. Let gel equilibrate in buffer for 15-20 minutes 6. While the gel is equilibrating, put transfer paper in methanol to wet 7. Pick transfer paper up with hands and put in transfer buffer 8. Make sandwich (wet sandwich with Transfer Buffer as needed) on transfer plate. a. First put down pad that is wetted in transfer buffer b. Put transfer paper on top(start putting it down in the middle to prevent bubbles) c. Put gel on d. Put wet pad on top e. Slightly compress with test tube—do not move sandwich 9. Mop up puddles on plate 10. Put the top on the plate and then attach plugs 11. Turn the current all the way down and the voltage all the way up. Put the voltage on 1X and the current on 10X. Set the current to 112.5 12. Transfer for 1HR 13. After transferring, make sure all the protein in transferred by making sure the marker on the gel has transferred 14. Put transfer paper in 25 mls of blocking solution for 1-2 hours at room temp or overnight at 4 degrees VII. Secondary 1. Incubate paper in secondary or HRP strepavidin for one hour at room temp (Dilute in 5% BSA in TBST) or at 37 degrees for ½ hour 2. Two quick washes in TBST, then 2 slow 10 minute washes in TBST VIII. Exposure 1. Cut transparency to fit in film developer 2. Put one squirt of each solution from the western blot analysis kit into a dish 3. With forceps put the transfer paper in the solution and wet 4. Put the transparency in the film developer and put the transfer paper in the transparency 5. Develop film using a film developer.