Figure S 1
advertisement

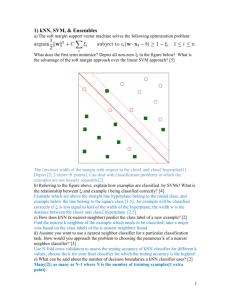
Figure S 1. Comparison of KNN models submitted to MAQC-II project. MCC performance comparison of all KNN models among four teams. All teams generally agree that endpoints E, H, and L are easy and result in high performance values. However, DAT18 tends to perform better than other groups for many of the endpoints: D, E, G, H, and I. Figure S 2. Parameter landscapes depicting the robustness of model selection. Each heatmap represents a two-dimensional slice of the four-dimensional parameter space (rank method, N, k, θ). In each case, the rank method is fold-change with p-value threshold of 0.05. The black ‘X’ indicates the peak performing model on cross-validation. The black triangle indicates the peak performing model on external validation. The thick blue contour line indicates the boundary of a region that does not perform significantly different from the peak cross-validation model with a p-value of 0.05. Figure S 3. Distribution of the difference between external and cross-validation. The kernel-smoothed estimate of the probability density for the difference between external validation and cross-validation of candidate models submitted to the MAQC-II project for all endpoints combined. The black circles indicate where the proposed KNNbased data analysis protocol performed for each endpoint. Figure S 4. Clinical utility of multiple myeloma and neuroblastoma predictive performance. Regardless of parameter selection, the KNN classifier predicts overall survival and event free survival of both the multiple myeloma and neuroblastoma datasets better than random chance. Plots represent distributions of average AUC and MCC external validation performance, with the MCC scaled to [0,1]. The negative controls are datasets with randomly permuted class labels and the positive controls are datasets with class labels corresponding to gender. P-values indicate the probability that a randomly selected model from the negative control performs better than a randomly selected model from the true dataset. Supplemental Data Table S 1. KNN data analysis protocol (kDAP). a: Proposed sensible KNN preanalysis data preparation protocol. b: Sensible KNN data analysis protocol. (a) Data sources and preparation Modeling Process Description Datasets Quality Control Gene Expression Calculation & Normalization Classifier Study The MAQC Consortium inspected the microarray data and removed low-quality chips. The MAQC Consortium distributed MAS5.0-calculated gene expression data. We evaluated alternate methods, but we abandoned batch-based calculation methods (e.g. PLIER and RMA) for reasons of clinical utility (i.e. patients do not always arrive in batches). We evaluated mean-centered and non-parametric quantile normalization methods, but these had no effect on classifier performance and did not remove noticeable batch effects. Data transformation We constructed our KNN models using training sets distributed by the MAQC Consortium. We tested the reliability of our final selected model using the multiple myeloma dataset from a different microarray platform, which was not part of the standard data distribution. For single channel chips, we used log2 of gene expression. For two channel chips, we used the log2 of the ratio: (sample intensity/reference intensity). We evaluated the data analysis protocols of four teams that used KNN. We also evaluated KNN models from biomedical literature. We categorized each model by the parameters that were varied, and studied the effects of each parameter before choosing the 6 parameters included in this study. (b) Data Analysis Plan: Cross Validation, Model Creation, and Performance Evaluation Analysis Step Explanation Step 1. 5-fold Crossvalidation Partition training data into two class groups. Randomly divide into 5 evenly-sized class sub-groups. Combine two sub-groups (one from each class) to yield 5 non-overlapping groups with similar prevalence to the original training data. Repeat steps 1.1-1.5 five times (folds) with each group held out once for testing. Step 1.1. Feature ranking Sensible methods for ranking features include a significance score and a minimum acceptable significance threshold*: o SAM: genes ranked by the significance analysis of microarrays (SAM) using delta=0.01 o FC&(P<0.05): Calculate statistical significance (P) of features using a simple two-sided t-test with unequal variance. Retain genes with P<0.05 and rank by fold change (FC). FC = 2^|mean(log2(class1)) – mean(log2(class2))|. o P&(FC>1.5): retain genes with FC>1.5 and rank by P. Other possible ranking methods include modified T-statistic or gene set enrichment analysis (GSEA). *Adjust the threshold if fewer than N genes pass (e.g., p’=p/1.5, FC’=FC2/3, delta’=0.001) Step 1.2. Feature selection Step 1.3. Classifier construction The size of the feature set (N) is always dataset-dependent. First, compare performance of feature lists of many sizes or use domain knowledge to determine a reasonable parameter space for the clinical problem. We used 5 to 200 features in increments of 5 plus a negative control set of all features passing the minimum threshold. For complex problems, feature set size over 200 may be worth exploring. Verify that the negative control feature set always underperforms the model selected using the reasonable range. Develop a cohort of classifiers for each feature set, varying dataset-dependent parameters over continuously-spaced ranges: o Determine a reasonable range for the number of neighbors, k, based on dataset size. We varied from 1 to 30* (significantly over the size of the smallest class used for training (positive J=22)). Values of k higher than 30 may be worth exploring for large, well-balanced datasets. o For dataset-independent parameters, use the most commonly-accepted from literature (for KNN, Euclidean Distance and equal-weight voting). *Typically, even values of k might lead to ties when using the conventional threshold of 0.5. In general, ties may occur when the threshold is chosen to be an integer ratio of k. To avoid ties, we choose a range of thresholds that do not include integer ratios of k (See Step 1.4). Step 1.4. Decision threshold Step 1.5. Test set prediction Evaluate classifier decisions across many thresholds. We used T=32 different thresholds, linearly spaced between 1/64 and 63/64. For KNN, we use a T that is one greater than a prime number greater than k. This avoids tied decisions. For a given k, there exist only k unique reasonable thresholds that can provide different classification performance. Due to the relationship between k and threshold it is possible to evaluate far fewer than 32 different thresholds per k. For simplicity of presentation, we evaluate all 32 thresholds in our analyses. Use every constructed classifier to predict the class of each sample in the test set. The predicted binary class for a sample is class 1 when (k1/k)>threshold, where k1 is the number of neighbors that belong to class 1; otherwise we predict class 0. Calculate performance metrics (AUC* and MCC) based on results of all samples in each fold of the test data. Other metrics of interest include accuracy, sensitivity, and specificity. Accuracy and MCC can be modified to incorporate unbalanced costs of false positives and false negatives. *AUC calculation is independent of threshold. Step 2. Summary of classification performance metrics Calculate mean classification performance as the mean metric across five folds. Repeat Steps 1 & 2 10 times and calculate mean and variance of performance metrics. Select a model based on an application-specific performance metric such as 0.5*AUC+0.25*(MCC+1) Use maximum mean cross-validation performance to select the candidate model. Step 3. Final model assessment Use the entire training set to train a “final KNN model” with the chosen parameters (ranking method, N, k, threshold). Predict the labels of blind validation data (e.g. new clinical samples). Assess performance using Min(CV,EV) to avoid rewarding a model that performs exceptionally well, but without predictability.