Chemical analyses Metals and organic pollutants in soft tissues of
advertisement

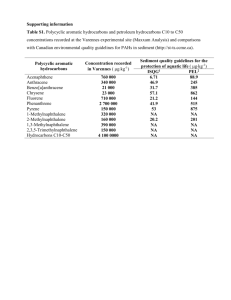
Chemical analyses Metals and organic pollutants in soft tissues of mussels were determined in freeze-dried, homogenized whole samples. Levels of Ni, Mn, Zn, Cu, Cd, Pb, Hg, As, and Pb were determined in acid-digested samples in a Perkin Elmer model Elan 6000 inductively coupled plasma mass spectrometer (ICP-MS) (Barata et al. 2005). Samples of similar weight of a certified reference material (dogfish muscle, NRCDORM-2; LGC Promochem, Middlesex, UK) were digested during each analytical run. Analyses of organic pollutants in mussel samples followed previous methods (Sánchez-Avila et al. 2011) and included fourteen Environmental Protection Agency (EPA) polycyclic aromatic hydrocarbons (PAHs), seven polychloro biphenils (PCBs), DDT derivatives and four altkylphenols (APs). PAHs were purchased from AccuStandard (New Haven, CT, USA) as a solution mix at 1000 μg L−1 in methanol; phthalates were from Supelco (Bellefonte, PA, USA) as a mix solution at 500 μg L−1 in methanol; PCBs (28, 52, 101, 118, 138, 153 and 180) were from Dr. Ehrenstorfer (Augsburg, Germany) as a mix solution at 10 μg L−1 in iso-octane; DDT derivates were also obtained from Dr. Ehrenstorfer GmbH; 4-Nonylphenol technical mixture (NP), 4-tertoctylphenol (OP), 4nonylphenol-mono-ethoxylate (NP1EO) and 4-nonylphenol-di ethoxylate (NP2EO) from Dr. Ehrenstorfer GmbH. Stock standard solutions (100 mg L−1) of each compound were prepared in absolute methanol and stored in the dark at −20 °C until use. These solutions were used to spike the mussel samples and draw the calibration curves. The surrogate standards (used for analyte quantification) for PAHs was a mix solution at 2000 μg L−1 in methanol containing anthracene D10, acenaphthylene D8, benzo(a)pyrene D12, benzo(h,g,i)perylene D12, fluoranthene D10, naphthalene D8, phenanthrene D10 and pyrene from Riedel-de Haën (Seelze, Germany), purchased as a solid; PCBs surrogate standard was PCB 209 from Dr. Ehrenstorfer GmbH, used as a solution at 10 μg L−1 in iso-octane; alkylphenols surrogate standard was 4-nnonylphenol-d8 and nonylphenol monoethoxylate D2 from Dr. Ehrenstorfer (Augsburg, Germany) as a solution at 100 μg L−1 in acetone; anthracene d10 used as internal standard, from Dr. Ehrenstrofer as a solution of 10 μg L−1 in cyclohexane. HPLC-grade methanol, hexane, dichloromethane and acetone for organic trace analyses were supplied by Merck (Darmstadt, Germany). Extraction and clean up: surrogate standards (4-n-NP-D8, NP1EO-D2, 4.4-DDD 13C12, PCB 209, PAHs surrogates solution were added to a sample aliquot of (0.1 – 1g), to get a final concentration of 100 ng mL−1. Samples were homogenized and kept at 4ºC overnight and subsequently extracted by sonication (10 min) one time with hexane/dichloromethane (1:1, v/v) in a ratio of 100 mL per gram of dried mass. The second part of the extraction (same ratio) was performed with a mixture of hexane/acetone (1:1, v/v). After each extraction step, samples were centrifuged for 10 min and the extracts were combined and concentrated to approximately 1 mL under a nitrogen current using a TurboVap LV system (Capiler Life Sciences, UK) at 25°C. Extracts were subsequently cleaned up by solid phase extraction (SPE) cartridges with 5 g of florisil, previously conditioned with 20 mL of hexane/dichloromethane (1:1,v/v) and 20 mL of hexane/acetone (1:1, v/v). The sample extract was eluted with 20 mL of hexane/dichloromethane (1:1, v/v) and 20 mL of hexene/acetone (1:1, v/v). The eluent was concentrated to a volume of less than 1 mL under a nitrogen current at room temperature and reconstituted with ethyl acetate to a final volume of 200 µL, with addition of 1 μg mL−1 of the internal standard Anthracene D10. Final extracts were analyzed by gas chromatography – electronic impact – mass spectrometry in tandem (GC-EI-MS/MS). Analyzes were performed using an Agilent 7890A GC System (Agilent Technologies, Palo Alto, CA, USA) interfaced to a 7000A triple quadrupole mass spectrometer system (Agilent, USA). Separation and detection method parameters were similar to those reported by (Sánchez-Avila et al. 2011). For increased sensitivity and specificity, peak detection and quantification was performed in Selected Reaction Monitoring (SRM) mode using two transitions per each compound. Internal standard quantification was performed using the surrogate standards method. Five points calibration curve were constructed at 100, 500, 1000, 5000 and 10000 ng/g with good linearity over this concentration range (R2 > 0.994). The method limits of detection (MLD) of the analytical method were calculated with spiked mussel samples at 100 ng/g of native compounds. MLDs for alkylphenols varied from 26.3 to 125.1 ng/g; for PAHs from 7.3 to 58.3 ng/g; for PCBs from 3.8 to 19.1 ng/g; for organochlorinated pesticides from 3.9 to 31.6 ng/g. Uncontaminated mussel tissue doped with 100ng/g of a solution containing all tested native compounds was analyzed along with samples to determine recovery rates. Sample blanks were rigorously performed to eliminate any external source of contamination.