Chapter 11 - Intermolecular forces of Solids and Liquids
advertisement

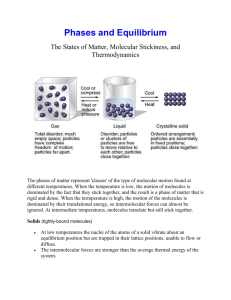
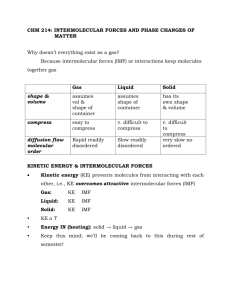
Chapter 11 - Intermolecular forces of Solids and Liquids For gases, we used the Kinetic Molecular Theory to explain observed behaviour of gases. This is adequate to explain the behaviour of collections of gaseous atoms or molecules since they are characterised by large intermolecular distances. Hence, at ordinary P and T, there are negligible attractive forces between gas molecules; gas molecules’ behaviour can be described independently of the actual chemical species note PV = nRT (n = # of gas particles only) Gas molecules low density, very compressible, whereas liquids and solids are not very compressible. Liquids the molecules move freely past each other liquids take the shape of their container, high density Solids the atoms or molecules are very rigidly held in place, with little freedom to move (long range order). Density of solids is usually higher than liquids (H2O big exception) Intermolecular Forces Intermolecular forces - are defined as the attractive forces between molecules (e.g. Hbonding, dipole-dipole etc). Note think of international between nations. Intramolecular forces - forces that hold atoms together in a molecule (e.g. chemical bonding). In general strength of intramolecular forces >>> strength of intermolecular forces. e.g., boil 1 mol of water 41 kJ of energy (overcoming intermolecular forces). H2O (l) H2 (g) + ½ O2 (g) rxnH = +286 kJ (overcoming intramolecular forces). Types of Intermolecular forces 1. dipole - dipole - act between polar molecules e.g. NF3 or HCl Note molecule must have dipole moments. electrostatic forces, obey Coulomb’s Law F q1q2/r2 2. Ion-dipole forces - occur between an ion (cation or anion) and a dipole. Hydration of an ionic salt NOTE: magnitude depends on charge and size of ion magnitude of dipole (c.f. Coulomb’s Law above). 3. Dipole-Induced Dipole forces - Let’s place a polar molecule (possesses a dipole) near a non-polar molecule. 2 - + dipole nonpolar species (symmetrical charge distribution) As we place the polar species near the nonpolar molecule, electron distribution of the nonpolar species becomes distorted. Hence, we have induced a dipole - + ------- ++++ ++++ ---- ++++ The ease of inducing a dipole is related to the “polarisability” of the non-polar system. Polarisability, the ease with which an electron distribution for a neutral molecule or atom can be distorted, depends primarily on the magnitude of the electron-nuclear attractive forces. These forces also depend, of course, on the magnitude (strength) of the dipole. Note polarisability increases with an increase in the molar mass of the atom or molecule. 4. Dispersion Forces - a bit more difficult to understand. These forces of attraction are also called London forces after Fritz London: Quantum Mechanical Treatment (1930). In a nonpolar molecule, the charge distribution is, on average, spherically symmetric. However, we realise that electrons are constantly in motion. Therefore, at any given time there may be an excess of electrons existing in one part of the atom or molecule. Hence, an instantaneous dipole is produced which will then proceed to distort the electron distribution of the neighbouring molecule, causing the atoms or molecules to be attracted towards one another. This force is significant only when the distances between the atoms or molecules is very small. The strength of dispersion forces also depends on ease of polarising the atom or molecule. Since the polarisability depends on size (mass) of atom or molecule, we should see an increase in the magnitude of the dispersion forces with increasing molecular mass. i.e., large atoms small atoms e’s far from nucleus less attractive forces easily distorted. e’s close to nucleus greater attractive forces hard to distort. Dispersion forces are present in all atoms and molecules. They are the only forces of attraction in nonpolar species. 3 NOTE non-polar molecules Cl2 Br2 gas liquid I2 solid Why? I2 and Br2 are more polarisable (i.e., the electron distributions are more easily distorted). Another example, look at the melting points of the following substances. CCl4 (-23.0C) < CBr4 (90.0C) < CI4 (171.0C) again due to an increase in the polarisability from Cl < Br < I Example What forces of attraction are present in these atoms and molecules? a) HCl b) Cl2 c) CCl4 HCl dipole-dipole and dispersion Cl2 dispersion CI4 dispersion (non polar molecule) Hydrogen Bonding Hydrogen bonding (H-bonding) is a very important, special type of dipole-dipole interaction between the H atom in a polar bond and an electronegative atom (N, O, F). e.g. water H2O O H H O H H O H H O H H Hbonds Covalent bonds Tetrahedral arrangement of H atoms around each O. These attractive forces are strong for 1. NH3 2. H2O 3. HF 4. Alcohols (e.g., methanol, CH3OH) 5. Amines (e.g., methyl amine, CH3NH2) 6. Carboxylic acids (e.g., CH3OOH) Structure and properties of water Properties (1) excellent solvent (due to dipolar) high heat capacity (due to intermolecular H-bonding) 4 Water (liquid) vs. water solid (ice) Liquid water exists as a “tetrahedrally” H-bonded system (alluded to earlier); solid water is also approximately tetrahedrally bonded (i.e., 2 covalent bonds and two hydrogen bonds). However, the density of water >>> density of ice. This is due to the rather open structure of water in the solid state (i.e., there is a hexagonal arrangement of water molecules in the solid state), compared to the more random structure of liquid water. The density of water actually increases from 0 4C. Note: when ice melts, there are a few molecules with enough kinetic energy to break from the H-bonded structure and they become trapped in the “cage-like” structures. Hence, we must consider two processes. 1. trapping of H2O molecules 2. thermal expansion From 0 4C trapping dominates, yielding an increase in the number of water molecules/unit volume (density increases slightly). After 4C thermal expansion dominates, and the more normal pattern of decreasing density with increasing temperature is observed. NOTE: SO FAR WE HAVE DEALT WITH ATTRACTIVE FORCES We also have repulsive forces (e- -e- and nuclear-nuclear); these forces became great when the distance between molecules is decreased. This is why we can’t compress easily liquids and solids. Surface Tension and Viscosity Surface Tension water beads on cars, glass etc., oil (grease) can form spots on a water surface (just look at the kitchen sink after you have washed away all the spaghetti sauce from the dishes). WHY? reason is surface tension definition the amount of energy needed to stretch or increase the surface by a unit of area IMPORTANT ENERGY IS NEEDED let’s look at the freshly waxed car. Water molecules have a great attraction for themselves but only have very weak attractive interactions with nonpolar substances like a wax (or air for that matter). 5 Water droplet Waxed car surface At the surface of the droplet, the water molecules are pulled inwards (downwards) and sideways by the H-bonding interactions (i.e., towards the centre of the droplet). At the surface, there is only a very weak attractive interaction between the water and the air. Note that the attractive interactions between the water and the waxed surface are also very weak. What shape provides the least surface contact? The sphere. Of course, gravity pulls the water droplet towards the surface. Attractive forces between like molecules are known as cohesive forces. Attractive fornces between unlike molecules (or between molecules and a solid surface) are known as adhesive forces. In the case of the water droplet on the waxed surface, the cohesive forces are significantly larger than the adhesive forces! n a glass capillary, however, water forms a atom adhesion vs. cohesion force Water vs. Hg in a capillary H2O in a capillary, adhesion > cohesion meniscus water pulled up along wall For Hg in a capillary, cohesion > adhesion depression occurs when a capillary tube is dipped in Hg. Viscosity The measure of the resistance of a fluid to flow. gases very low viscosity liquids variable e.g., benzene has a low viscosity, whereas molasses has a high viscosity. Intermolecular forces are very important! 6 e.g. ethylene glycol (antifreeze) H H H C C H OH OH Intermolecular H - bonds attractive forces high, therefore the viscosity is high. benzene, Characterised by dispersion forces and it is a planar, flat molecule viscosity is low. It is relatively easy for the benzene molecules to slide past one another What about the following series? hexane octane decane CH3CH2CH2CH2CH2CH3 CH3(CH2)6CH3 CH3(CH2)8CH3 0.326 * 10-2 g/cms 0.542 * 10-2 g/cms 0.92 * 10-2 g/cms WHY? Two reasons (1) molar mass increases increasing dispersion forces. In addition, as the chain lengths increase, they become entangled! The ease of entanglement increases with increasing chain length. Viscosity as T . WHY? H2O as example. T , H-bonds break, attractive forces decrease viscosity decreases. Changes of State evaporation molecules have enough kinetic energy to escape form the surface. condensation as vapour pressure increases, K.E. goes down slightly for some molecules and more return to the liquid state When the rate of evaporation = rate of condensation, we have obtained the equilibrium vapour pressure. Vapour pressures and intermolecular forces Note that molecules in the liquid phase escape from the surface of the liquid to the vapour phase. Only a few molecules will have a sufficient amount of kinetic energy to overcome the intermolecular forces present in the liquid phase to escape to the vapour phase. 7 vapour liquid Hence, the magnitude of the vapour pressure of the liquid is an indication of the strength of intermolecular forces. If the intermolecular forces are strong, the substance has a low vapour pressure; if the intermolecular forces are weak, the substance will have a high vapour pressure. Note that there is an inverse relationship between intermolecular forces and vapour pressure. How do vapour pressures vary with temperature? Vapour pressures increase with increasing temperature. As T increases, the kinetic energy increases, and hence, more molecules are able to overcome the intermolecular forces present at the surface of the liquid and escape into the vapour phase. Molar heat of vaporization H2O (l) H2O(g) rxnH vapH, molar heat of vaporisation. (energy required to vaporize 1 mole of liquid). The enthalpy of vaporisation is an excellent indication of the strength of the intermolecular forces. Note magnitude of vapH is large, strong intermolecular attractive forces, whereas if the enthalpy of vaporisation is small, the intermolecular forces are weak. How do we determine vapH? Use the Clausius-Claypeyron equation lnP = -vapH/RT + C (linear equation); P = vapour pressure, T = temp in K, and C is a constant. look at two different temperatures (T1 and T2) lnP1 = -vapH/RT1 + C lnP2 = -vapH/RT2 + C lnP2/P1 = -vapH/R (1/T2 - 1/T1) Hence, a plot of ln P vs. 1/T shuld be linear with a slope equal to -vapH/R. Example The vapour pressure of water at 308 K is 42.18 mm of Hg. Given that the vapH of H2O is 40.79 kJ/mol, calculate the vapour pressure of H2O at 338 K. 8 n n P2 vap H 1 1 = * - P1 R T 2 T1 P2 40.7 kJ/mol * 1000J/kJ 1 1 = * K P1 338K 308K 8.314 J mol n 4.10 = P2 P = 1.41 e -1.414) = 1 P1 P2 P2 P 2 = 42.18 mm Hg 4.10 P1 = 173 mm of Hg ANS: Boiling Points and Intermolecular Forces What about the point at which P2 = the external pressure? This is defined as the boiling point of the solution. Hence, boiling points are dependent on the external pressure (boiling points generally decrease with decreasing external pressure). When the external pressure is 1.00 atm, we define this temperature as the normal boiling point, which for water is exactly 100.0C or 373.15 K at 1 atm pressure. Hence, normal boiling points as well as vapH values give an indication of the strength of intermolecular forces. Note strong intermolecular forces high enthalpies of vaporisation and high boiling points. If the intermolecular forces are weak, the normal boiling points as well as the enthalpies of vaporisation are small. Examples Ar and CH4 and ethane have low b.p.s. (dominated by dispersion forces) Change ethane to ethanol ethanol has strong H-bonding, whereas ethane is characterised only by dispersion forces. Ethane boils at -88.6C; ethanol boils at 78.5C. Critical Temperatures and Critical Pressures Critical pressure - the minimum pressure that has to be applied to liquefy the gas at the critical temperature (or highest temperature where the liquid substance can exist) Critical temperature - the temperature above which a gas will not liquefy no matter how great 9 the pressure. e.g. CO2 familiar with two forms, the solid (known as ‘dry ice’) and the gas (atmospheric CO2). Liquid CO2 cannot be made at atmospheric pressure (note CO2 has a Pc = 73 atm). Supercritical CO2 ( i.e., CO2 at a temperature above its Tc = 31.0C) is used to decaffeinate coffee!! Solid liquid m H2O (s) H2O (l) fusH molar heat of fusion energy required to convert 1 mole of water form the solid to liquid state. Note fusH << vapH . This has to do with the differences in intermolecular distances (packing of molecules) in the various states. When we heat a solid, the molecules are packed close together. As we progress to the liquid state, the molecules are still fairly close together (i.e., strong attractive forces). However, as we progress to the gaseous state, we find the intermolecular distances are large and the molecules are far apart (therefore, weak interactions). Heating/Cooling Curves - graphically show the heating, melting, and vaporising processes. Note that the melting points and the boiling points have invariant (constant) temperatures. Solid vapour m CO2 (s) ⇌ CO2 (g) subH molar heat of sublimation energy required to convert 1 mole of the substance from the solid to gaseous state. The reverse process (where the gas directly to the solid state is known as deposition). Good examples, CO2 at atmospheric pressure, I2(s) Note subH = fusH + vapH from Hess’s Law Example Calculate the heat required to evaporate 37.3 g of H2O at its boiling point. Process H2O (l) ⇌ H2O (g) g of H2O = 37.3 moles of H2O = 37.3g * 1 mol/18.02g = 2.07 moles H2O energy (heat) needed = 2.07 moles of H2O (l) * 40.79 kJ/mol = 84.2 kJ of heat vapH = 40.7 kJ/mol Phase Changes Transform one phase to another, e.g., ice water is an example of a solid liquid phase change (needs heat!!). 10 Phase Diagrams give the overall relationship among the solid, liquid, and vapour phases at many temperatures and pressures. Look at some typical phase diagrams. external pressure (usually atm) y axis; T(C) x-axis water 1. Below OC generally, solid (note the pressure dependence of the melting point of water). 2. At 100C. Intersect the liquid-vapour m line, p = 1 atm exactly. (note the pressure dependence of the boiling point discussed previously). 3. All three curves intersect at a specific point called the triple point. This is a very important point as it is the only point where all three phases are in m with one another. For water the triple point is at 0.006 atm and 0.01C. 4. Note that the solid-vapour m exists only below the triple point. 5. What happens if we lower the external pressure to 0.10 atm? 6. m.p. increases, b.p. decreases very important look at CO2 phase diagram 1. The liquid region (phase) lies well above atmospheric pressure as the triple point for CO2 is above atmospheric pressure (5.2 atm and -57C). 2. The solid-liquid boundary line has a positive slope (i.e., melting point increases with increasing pressure). Compare to water. 3. Note the critical point for CO2 and compare with water. Crystal Structure crystalline solid - rigid and long range order amorphous solid - lacks a long-range 3-D structure In a crystalline solid - atoms, molecules or ions occupy specific positions. Arranged so that the net attractive forces are a maximum (lowest energy state). the centre of the positions occupied by atoms (molecules or ions) lattice point unit cell - basic repeating arrangement of the atoms, molecules or ions. There are seven types of unit cells. The crystal lattice is a 3-D array of points, each rep. identical environment in the crystal. 1. cubic a = b = c; = = = 90 2. tetragonal a = b c; = = = 90 3. orthorhombic a b c; = = = 90 4. rhombohedral a = b = c; = = 90 5. monoclinic a b c; = = 90 90 6. triclinica b c; 90 7. hexagonal a = b c; = = 90 = 120 11 look at a cubic unit cell simple cubic body-centred cubic face-centred cubic NOTE each atom is shared by a number of unit cells. example simple cubic cell vs a face-centred cube (Slide). Examples of Atoms and Molecules with differing simple unit cells gold face centred cubic cell tungsten body-centred cubic cell NaCl ionic salt face centred cubic structure Packing Spheres closest packing refers to the most efficient arrangement of spheres try to imagine the difference packing golf balls in a large box (more eff golf balls fill holes) Two Types 1. ABA hexagonal close – packing (hcp) 2. ABC cubic close - packing (ccp). Note in hcp the spheres in alternate layers occupy the same vertical positions, whereas in ccp, for every fourth layer, the spheres occupying the same vertical space. For both structures each sphere is surrounded by 6 other spheres in the same layer, 3 spheres in the layers immediately above and below it. The co-ordination # = 12 Coordination # the # of atoms (or ions) surrounding an atom (or ion) in the crystal lattice. Compare with a simple cubic cell (scc). co-ordination # = 8 (8 nearest neighbours) scc has a lower packing efficiency i.e., the hcp and ccp “plug” the holes better What can we learn about an atom from its crystal structure? Very important we can calculate the atomic radius! Example 1. Simple cubic system Simple cube a = b = c; Length of edge a = 2r if we know a we can calculate the atomic radius. 2. Body Centred Cube. b2 = 2a2; b = 2 a; c2 = a2 + b2; c2 = a2 + 2a2 = 3a2 12 c = 3 * a; a = c/ 3; but c = 4r; a = 4r/ 3 3. Face Centred Cube 1 = 8 r cubic unit structure cell sides are equal c = 4r c2 = 16r2 = a2 a = 8 r Types of Crystals 1. Ionic e.g. NaCl 2. covalent e.g. SiO2 3. molecular e.g. I2, H2O (icc), sucrose 4. metallic e.g. Au, Ag, Ni ... Types of Crystals and General Properties Type Units at Lattice Points Forces General Properties Examples (1) Ionic “+” and “-“ ions electrostatic high m.p., hard doesn’t conduct heat and electricity well NaCl CsCl NaI (2) Covalent Atoms covalent bonds hard, high m.p., doesn’t conduct heat and electricity SiO2 diamond graphite molecular molecules or atoms; dispersion, dipole-dipole H-bonds; soft, low m.p., poor conductor of heat and electricity, e.g., I2, H2O, sucrose metallic Atoms; metallic bonds; soft hard, low to high m.p.s, good conductor of heat and electricity, e.g., Ni, Al, Ti, etc Amorphous Solids lack a regular 3-D arrangement. Best-known example glasses Glasses are optically transparent fusion products of inorganic materials that have cooled to a rigid state without recrystallising. Glasses have some liquid and some solid properties Coloured glasses due largely to the presence of metal ions. e.g. green glass - iron oxide (Fe2O3) yellow glass UO2 blue glass Co(II) and Cu(II) oxides