Comparing Extracellular Secretion Mechanisms in E. Coli Strains
advertisement

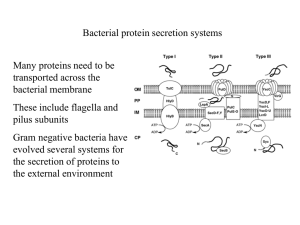
Comparing Extracellular Secretion Mechanisms in E. Coli Strains BL21(DE3) and Nissle 1917 Cornell University Department of Biological and Environmental Engineering Author: Matthew S. Russell, M. Eng. ‘09 Faculty Adviser: John C. March, Ph. D. Date Submitted: ________________________________ Matthew S. Russell: ___________________________________________ Date: ____________________ Abstract: An important element of engineering probiotic bacteria for the treatment or prevention of disease is the organism’s ability to secrete useful concentrations of a drug, usually a protein produced from recombinant DNA. Achieving useful concentrations of secreted proteins in the gut of a patient may require maximizing secretion by a specific pathway or combining more than one pathway within a probiotic bacterium. In order to make informed decisions about how to attain desired secretion rates and amounts from a probiotic bacterium, we must understand the relative efficiencies and limitations of different secretion pathways within a specified probiotic bacterium. To this effect, this work examined the relative efficiencies of histidine-tagged glucagon-like peptide-1 (Glp1) secretion from two strains of Escherichia coli, Nissle 1917 and BL21(DE3), using a modified alpha-hemolysin (HlyA) secretion system and using a secretion tag for flagellin (FliC) [1,2]. These two secretion systems have been developed by Lee et al. and Majander et al. respectively. We looked into what effect controlling the HlyA secretion system from different promoters would have using PLac, an inducible expression promoter, and PT7, a promoter that recruits a high efficiency RNA polymerase. Preliminary results show that the HlyA secretion system is more effective in Nissle 1917 than the FliC system. They also show that controlling expression of secreted protein from PT7 increases secretion relative to control from PLac in BL21(DE3). Introduction: Probiotics offer an exciting opportunity for treatment and prevention of disease because they can be administered orally, live in the gut for some time, and respond to environmental stimuli. However, they must be capable of delivering useful amounts of drugs. Let us assume an average lumen volume of the human small intestine of 393 ml [3], and that a probiotic bacterium is able to colonize this volume to an optical density at 600 nm of 1.0 (OD600), or approximately 1*109 colony forming units per ml. Estimates for the secretion capacity of the recombinant fusion proteins using the HlyA secretion pathway are 3-5% of total cell protein, or approximately 2.3 mg/L/OD [4,1]. Then, assuming that this is the amount of protein drug absorbed by or delivered to intestinal epithelial cells (IECs) in a day, since these estimates are reported without mention of a time period, the HlyA pathway is only capable of delivering a maximum of 0.90 mg of drug per day. In reality it is unlikely that a probiotic bacterium will be able to colonize the entire lumen of the small intestine to this extent since it will have to compete with other native microorganisms that are likely better suited to this environment. Even with the increased secretion rate reported by Lee et al. for a HlyA hypersecretion mutant of 9.2 mg/L/OD, the above assumptions yield only 3.6 mg of protein drug absorbed by or delivered to IECs per day [1]. This amount of drug delivery may be sufficient for treatment or prevention of some diseases, but in order for probiotics to become broadly applicable, they must be capable of secreting increased amounts of recombinant proteins to the intestinal lumen. This study investigated secretion of fusion proteins formed by combining his-tagged Glp1 with the secretion tags for HlyA and FliC secretion. Glp1 was chosen because of its utility in other experiments in our lab. The HlyA and FliC secretion pathways were chosen because both are pathways that translocate protein across the inner and outer membranes of Gram-negative E. coli and into the extracellular medium. This is an important feature for probiotic design because extracellular medium is analogous to the lumen of the gut in vivo, and secreted proteins must reach this space to contact and act on IECs. Other secretion systems exist that transport proteins into the periplasm, but few are capable of moving proteins past the outer membrane. The HlyA secretion system is an example of type one secretion in E. coli. Type one secretion systems include an ATP binding cassette protein, or hemolysin B (HlyB) in the HlyA system, which binds a 60 AA signal sequence at the C-terminus end of HlyA. They also include a membrane fusion protein, hemolysin D (HlyD), which connects the inner and outer membranes, and an outer membrane protein (TolC). Once HlyB binds the signal sequence of a target protein, it passes its protein target into the pore formed by HlyB and TolC and allows the target protein to exit the cell [5]. HlyA, the primary target of this secretion system, is a hemolytically active virulence factor. It is activated by hemolysin C (HlyC) after translation by acylation of its N-terminus [1,6,7]. Since our experiments use only a 219 AA portion of the HlyA protein, and we do not include the gene for HlyC in our constructs, secreted proteins should not display hemolytic activity. However, this claim must be verified before consideration for in vivo testing. DNA encoding this signal sequence was engineered to produce a hypersecretion phenotype by inserting rare codons and decreasing transcript copy number [1]. The FliC secretion system used is an example of a type three secretion system in E. coli. All type three secretion systems are homologues of a flagellar basal body, which spans the inner and outer membranes of E. coli and forms a molecular “syringe” through which transported proteins can travel. The structure, assembly and function of the flagellum requires over 50 genes in E. coli and is not discussed in detail here [2,8]. Flagellin (FliC) is the major constituent of the flagellum. FliC contains an N-terminus signal sequence which is recognized by proteins in the basal body and permits its transport through the filament. A capping protein (FliD) then assembles FliC monomers into the filament at the end of the flagellum [2,9]. Mutant strains, where FliD does not function correctly, allow secretion of FliC monomers into extracellular media [9]. With this knowledge, Majander et al. were able to design a secretion system consisting of an E. coli strain with FliC and FliD deletions in the chromosome, and fusion proteins bound to the protein product of a 183 bp section of DNA coding for the FliC promoter, 5’UTR, and N-terminus of FliC [2]. Using this system, they report protein yields in the extracellular medium ranging from 1-15 mg/L. In previous work, we have been unable to achieve similar yields to those reported using this system. The organisms used in this study do not include chromosomal deletions of FliC and FliD, so we expect somewhat decreased yields from those reported due to competition between our secretion protein and wild type FliC. The probiotic bacterium chosen for this study was Nissle 1917 (Ardeypharm, Germany). This organism was chosen because of its utility in other experiments in our lab. Nissle 1917 has been shown to have anti-inflamatory effects on IECs and has been used and studied for treatment of inflammatory bowel disease and ulcerative colitis [10]. The mechanism of Nissle 1917’s anti-inflammatory effects is still not fully understood, but it works through a secreted factor, since contact with IECs is not required [10]. BL21(DE3) was also used for expression of secreted proteins because it expresses the T7 RNA polymerase, a high efficiency polymerase used for gene expression [11]. The goal of this work was to compare the HlyA and FliC secretion pathways in Nissle 1917 and BL21(DE3) and to evaluate the effects of controlling the production of protein designed for secretion via the HlyA system using the Lac and T7 promoters (PLac and PT7). PLac is a well established, isopropyl-β-Dthiogalactopyranoside (IPTG) inducible promoter, and PT7 is a promoter which recruits the high efficiency T7 RNA polymerase. We wanted to see if using PT7 would increase the amount of secreted protein, presumably by increasing transcript copy number, or if it would simply remove the hypersecretion phenotype as the findings of Lee et al. suggest [1]. Methods: Bacterial strains, growth and induction conditions: Bacterial strains used in this report are indicated in Table 1. Strains were grown in Luria-Bertani (LB) medium and on LB medium-agar plates with appropriate antibiotics. Liquid cultures were grown at 37°C with shaking at 250 rpm. However when protein secretion was induced, cultures were grown at 37°C without shaking (appropriate antibiotic and inducer concentrations were used here). Single colonies, or stocks prepared by growing single colonies and storing in 30% (v/v) Glycerol LB medium at 80°C, were inoculated into LB medium. PLac constructs were induced with 1.0 mM IPTG before incubation at 37°C. Antibiotics were added at the following concentrations: ampicillin, 100 μg/ml; chloramphenicol, 65 μg/ml; tetracycline, 25 μg/ml. DNA Constructs, Oligonucleotides, Plasmids, and Cloning: Table 1 displays a list of plasmids used here. DNA manipulations, cloning, and PCR were performed using standard protocols [12]. Cloning was performed in E. coli DH5α before plasmid recovery and transformation into expression organisms BL21(DE3) and Nissle 1917. Oligonucleotides were synthesized by IDTDNA and are shown in Table 2. The DNA sequence of the HlyA hypersecretion mutant was amplified from pLG612-1SD [13]. The DNA sequence of Glp1 was amplified from pFliCGlp1. The DNA sequence illustrated in Figure 2 was prepared using standard cloning techniques [12]. This sequence was inserted into pBluescript SK (Stratagene) and pivex2.3d (Roche) under control of PLac and PT7 respectively. Sequencing analysis was performed to confirm that constructs were mutation free using sequencing primers M13F and M13R. Vectors pBsGlp1HlyA and pivexGlp1HlyA were transformed into BL21(DE3). Vector pBsGlp1HlyA was transformed into Nissle 1917. Vector pvdl9.3 was transformed into all HlyA secretion constructs. Table 1. Strains and plasmids used. Strain or Plasmid Strains: E. coli DH5α Relevant Characteristics F- endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG Φ80dlacZΔM15 Δ(lacZYA-argF)U169, hsdR17(rK- mK+), λ– F– ompT gal dcm lon hsdSB(rB- mB-) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) E. coli BL21(DE3) E. coli Nissle 1917 E. coli Nissle 1917 PFliC-FliC-Glp1 Contains vector with 183 bp sequence containing PFliC, 5'UTR, N-terminus secretion sequence, and his-tagged Glp1 E. coli Nissle 1917 PLac-Glp1-HlyA E. coli BL21(DE3) PLac-Glp1-HlyA E. coli BL21(DE3) PT7-Glp1-HlyA Contains pBsGlp1HlyA, pvdl9.3 Contains pBsGlp1HlyA, pvdl9.3 Contains pivexGlp1HlyA, pvdl9.3 Plasmids: pBluescript SK pivex2.3d pLG612-1SD pFliCGlp1 pvdl9.3 pBsGlp1HlyA pivexGlp1HlyA Source PLac Expression Vector PT7 Expression Vector Vector containing HlyA hypersecretion phenotype DNA Vector containing his-tagged Glp1 Vector containing HlyB, HlyD pBluescript SK containing his-tagged Glp1 and 219 AA Hly A secretion sequence pivex2.3d containing his-tagged Glp1 and 219 AA Hly A secretion sequence Table 2. Table of oligonucleotides used. Oligonucleotides Sequence 5’ ATTGCGGCGCTATGCATCATCATCATCAT 3’ 5’ ATTGCGGCCGCATGCATCATCATCATCAT 3’ 5’ GCCACTAGTTCCTCGGCCTTTCACCAG3’ 5’ GCCACTAGTGGAAATTCTCTTGCTAAA 3’ 5’ ATTCTCGAGTTATGCTGATGCTGTCAA 3’ M13F M13R Description Glp1 Forward (for pBluescript SK) Glp1 Forward (for pivex2.3d) Glp1 Reverse HlyA Forward HlyA Reverse M13F Sequencing primer M13R Sequencing primer Faping Duan [14] Faping Duan, Benita WesterlundWikström [2] This work This work This work Stratagene Roche Victor de Lorenzo [13] Faping Duan Kelvin Lee [1] This work This Work Figure 1. Diagram of Glp1-HlyA DNA sequence. Protein recovery and analysis techniques: Protein recovery techniques were adapted from Majander et al. and used to collect and analyze secreted and cellular protein from different secretion constructs [2]. Bacteria were grown in 3 ml of LB medium overnight as previously described. OD600 was taken and 1*108 cells, or 0.100 ml of culture at OD600=1, was added to 15 ml of LB medium. This was grown overnight for 18 hours, then OD600 was taken and samples were divided into 13 and 1.5 ml fractions. 1.5 ml fractions were spun at 10000g for 10 minutes, 4°C to pelletize cells, 13 ml fractions were spun at 2000g for 30 minutes, 4°C to pelletize cells. Supernatant was poured off of 1.5 ml fractions and 40 μl of BugBuster Master Mix (Novagen) and phenylmethanesulphonylfluoride (PMSF) were added to recover intracellular protein. Once intracellular protein recovery was completed, according to BugBuster instructions, 10 ul of 5X SDS loading buffer was added and samples were stored at 4°C. Meanwhile supernatant from the 13 ml fractions was recovered and protein was precipitated with 10% trichloroacetic acid (TCA) for 30 minutes on ice. PMSF was also added. Protein was spun down at 16000g for 15 minutes, 4°C. Then Supernatant was poured off, and protein was washed twice with 50% (v/v) Ether, 50% (v/v) EtOH and re-pelletized. Finally recovered protein was dissolved in 25 μl sterile MilliPore H2O, 25 μl 5X SDS loading buffer. Sodium dodecyl sulfatepolyacrylamide gel electrophoresis (SDS-PAGE) was performed loading a volume of extracellular secreted protein equivalent to the volume of medium containing 6.5*109 cells and a volume of intracellular protein equivalent to that from 7.5*108 cells (8.667 times as much secreted volume as cell volume). Standard western blotting techniques were used to transfer protein to a polyvinylidene fluoride (PVDF) membrane (Millipore), block it with 5% (w/v) milk, and incubate it with first anti-his, then antimouse. Western blots were developed with horseradish peroxidase and exposed to x-ray film for 15 minutes. Western blots comparing intracellular and extracellular protein from Nissle 1917 with the FliCGlp1 construct and with the PLac-Glp1-HlyA construct were obtained. A western blot comparing intracellular and extracellular protein from BL21(DE3) with the PLac-Glp1-HlyA construct and with the PT7Glp1-HlyA construct was also obtained. Results and Discussion: Preliminary results indicate that the HlyA secretion system is more effective and efficient than the FliC secretion system in Nissle 1917. Figure 2 shows the western blot results for cellular and secreted protein in Nissle 1917. However, the results for the FliC system were taken from a different western blot than those for the HlyA system. This is less than ideal and it would be better to see side- by-side results for these two systems on the same western blot to minimize the possibility of experimental error. The HlyA system, under control of PLac, does appear to secrete more protein overall than the FliC secretion system in Nissle 1917. Future work in Nissle 1917 will involve checking the reproducibility of these preliminary results expressing the T7 RNA polymerase in this organism so that we can compare secreting via the HlyA system under control of PT7 and PLac. Figure 2. Western blot results for cellular and secreted protein in Nissle 1917 using the HlyA and FliC secretion pathways. Figure 3 shows a comparison of secreted protein from BL21(DE3) using the HlyA system under control of PLac and PT7. Cellular protein fractions did not produce a signal. These results are taken from the same western blot, so there is less possibility of experimental error, but they still are preliminary and need to be tested for reproducibility. It has been suggested that laboratory strains of E. coli are deficient in their secretion pathways [15]. This may be why there is less signal seen for secreted protein in BL21(DE3) than in Nissle 1917 and there is no signal from cellular protein. These results seem to contradict those of Lee et al. who indicate that transcript copy number is inversely correlated with HlyA secretion since there is increased secretion when expression is controlled by a high efficiency promoter and RNA polymerase. However, other factors may come into play here such as transcript copy number and the expression strain. Further work is needed to understand the effects of controlling HlyA secretion with PT7. Figure 3. Western blot results for secreted protein in BL21(DE3) using the HlyA secretion pathway under control of PLac and P7. Works Cited: [1] Lee, Pat S., and Kelvin H. Lee. "Engineering HlyA Hypersecretion in Escherichia coli Based on Proteomic and Microarray Analysis." Biotechnology and Bioengineering 89 (2005): 195-205. [2] Majander, Katariina, Lena Anton, Jenni Antikainen, Hannu Lang, Mirko Brummer, Timo K. Korhonen, and Benita Westerlund-Wikstrom. "Extracellular secretion of polypeptides unsing a modified Escherichia coli flagellar secretion apparatus." Nature Biotechnology 23 (2005): 475-81. [3] Mann, Sabine, Pierre-Olivier Droz, and Marie Vahter. "A Physiologically Based Pharmacokinetic Model for Arsenic Exposure: II. Validation and Application in Humans." Toxicology and Applied Pharmacology 140 (1996): 471-86. [4] Blight, Mark A., and I. B. Holland. "Heterologous protein secretion and the versatile Escherichia coli haemolysin translocator." Trends in Biotechnology 12 (1994): 450-55. [5] Binet, R., S. Letoffe, J. M. Ghigo, P. Delepelaire, and C. Wandersman. "Protein secretion by Gram-negative bacterial ABC exporters - A review." Gene 192 (1997): 7-11. [6] Lim, Kheng B., Carthene R. Bazemore Walker, Lin Guo, Shahaireen Pellett, Jeffrey Shabanowitz, Donald F. Hunt, Erik L. Hewlett, Albrecht Ludwig, Werner Goebel, Rodney A. Welch, and Murray Hackett. "Escherichia coli α-Hemolysin (HlyA) Is Heterogeneously Acylated in Vivo with 14-, 15, and 17-Carbon Fatty Acids." The Journal of Boilogical Chemistry 275 (2000): 36698-6702. [7] Stanley, Peter, Vassilis Koronakis, and Colin Hughes. "Acylation of Escherichia coli Hemolysin: A Unique Protein Lipidation Mechanism Underlysin Toxin Function." Microbiology and Molecular Biology Reviews 62 (1998): 309-33. [8] Chilcott, Gavin S., and Kelly T. Hughes. "Coupling of Flagellar Gene Expression to Flagellar Assembly in Salmonella enterica Seovar Typhimurium and Escherichia coli." Microbiology and Molecular Biology Reviews 64 (2000): 694-708. [9] Minamino, Tohru, and Keiichi Namba. "Self-Assembly and Type III Protein Export of the Bacterial Flagellum." Journal of Molecular Microbiology and Biotechnology 7 (2004): 5-17. [10] Kamada, Nobuhiko, Kenichi Maeda, Nagamu Inoue, Tadakazu Hisamatsu, Susumu Okamoto, Kyong Su Hong, Takaya Yamada, Noriaki Watanabe, Kanji Tsuchimoto, Haruhiko Ogata, and Toshifumi Hibi. "Nonpathogenic Escherichia coli Strain Nissle 1917 Inhibits Signal Transduction in Intestinal Epithelial Cells." Infection and Immunology 76 (2008): 214-20. [11] Studier, F. W., and Barbara A. Moffatt. "Use of Bacteriophage T7 RNA Polymerase to Direct Selective High-level Expression of Cloned Genes." Journal of Molecular Biology 189 (1986): 11330. [12] Sambrook, Joseph, and David W. Russell. Molecular Cloning A Laboratory Manual (3-Volume Set). New York: Cold Spring Harbor Laboratory P, 2001. [13] Fernández, Luis A., Isabel Sola, Luis Enjuanes, and Victor De Lorenzo. "Specific Secretion of Active Single-Chain Fv Antibodies into the Supernatants of Escherichia coli Cultures by Use of the Hemolysin System." Applied Environmental Microbiology 66 (2000): 5024-029. [14] Duan, Faping, and John C. March. "Interrupting Vibrio cholerae Infection of Human Epithelial Cells with Engineered Commensal Bacterial Signaling." Biotechnology and Bioengineering 101 (2008): 128-34. [15] Pugsley, A. P., and O. Francetic. "Protein secretion in Escherichia coli K-12: dead or alive?" Cellular and Molecular Life Sciences 54 (1998): 347-52.