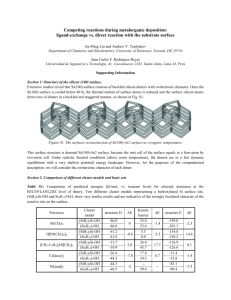
Supporting Material S1. A test calculation using B3LYP method with
advertisement

Supporting Material S1. A test calculation using B3LYP method with the all-electron basis set To confirm the reliability of the used effective core potential (ECP) basis set (LANL2DZ) on the Fe atom in our calculation, we performed an additional test calculation by using the all-electron basis set (6-311+G(d,p)) on the Fe atom. The 6-311+G(d,p) basis set and the LANL2DZ basis set were employed respectively to calculate the [Fe(H2O)n]2+ clusters with up to n=6, and the obtained average hydration energies are displayed in figure S1. As can be seen from figure S1, the trend of the average hydration energy obtained at the level of the LANL2DZ basis set is quite similar to that at the level of the 6-311+G(d,p) basis set. When n=6, the average hydration energy, 2.288 eV, from the calculation of the 6-311+G(d,p) basis set is very close to the value, 2.285 eV, from the calculation of the lanl2dz basis set. The exceptional case at n=1 does not affect the whole trend of the energy curve. So, with consideration of the time-consuming, it is reasonable to use the ECP basis set for our The average hydration energies (eV) calculations. 4.0 3.8 3.6 3.4 3.2 3.0 2.8 2.6 2.4 2.2 the 6-311+G(d,p) basis set the lanl2dz basis set 1 2 3 4 5 6 The number of water molecules (n) Figure S1. The average hydration energy calculated at the level of the 6-311+G(d,p) 1 basis set and the LANL2DZ basis set respectively S2. The average Fe-O distances evaluated by using B3LYP, MP2, B3PW91, and HF methods Since the structural feature including the bond lengths is also a factor to reflect the suitability of the used computational method, we thus summarized the average Fe-O distances obtained at the levels of B3LYP, MP2, B3PW91 and HF. As displayed in figure S2, the difference of the average Fe-O distances resulted from B3LYP and B3PW91 levels is tiny, just within 0.003-0.009 Å only. Meanwhile, the trend of the average Fe-O distances with the cluster size, obtained at the levels of B3LYP and B3PW91 respectively, are nearly same too. This also implies that both the B3LYP functional and the B3PW91 functional are reliable to describe the interactions in the current studies of the hydrated ferrous ion clusters. So, it is suitable to handle the The average distances of Fe-O (Ang) concerned systems by using the B3LYP functional. 2.20 B3LYP MP2 B3PW91 HF 2.15 2.10 2.05 2.00 1.95 1.90 1 2 3 4 5 6 The number of water molecules (n) Figure S2. The average Fe-O distances obtained at the levels of B3LYP, MP2, B3PW91, and HF, respectively. S3. The spin contaminations The disadvantage of unrestricted calculations is that the wave function is no longer an eigenfunction of the total spin, <S2>, thus some error may be introduced into the 2 calculation. This error is called spin contamination. If there is no spin contamination, the expectation value of the total spin, <S2>, should equal s(s+1), where s is 1/2 times the number of unpaired electrons. One rule of thumb which was derived from experience with organic molecule calculations is that the spin contamination is negligible if the value of <S2> differs from s(s+1) by less than 10%. The values of <s2> and s(s+1) in the calculations are displayed table S1. As can be seen from table S1, these spin contaminations are very weak for our investigated system and can be negligible. Table S1. The values of spin contaminations for the [Fe(H2O)1-19]2+ complexes. n 1 2 3 4 5 6 7 8 9 10 s(s+1) 6 6 6 6 6 6 6 6 6 6 <S2> 6.0036 6.0039 6.0038 6.004 6.0037 6.0034 6.0037 6.0037 6.0037 6.0038 n 11 12 13 14 15 16 17 18 19 s(s+1) 6 6 6 6 6 6 6 6 6 6.004 6.004 6.004 6.004 <S2> 6.0039 6.004 6.004 6.004 6.004 3