Exploring the chemical reactions leading to contamination during
advertisement

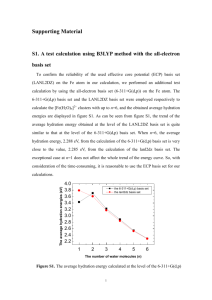
Competing reactions during metalorganic deposition: ligand-exchange vs. direct reaction with the substrate surface Jia-Ming Lin and Andrew V. Teplyakov Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716 Juan Carlos F. Rodriguez-Reyes Universidad de Ingeniería y Tecnología, Av. Cascanueces 2281, Santa Anita, Lima 43, Perú Supporting Information Section 1: Structure of the silicon (100) surface. Extensive studies reveal that Si(100) surface consists of buckled silicon dimers with zwitterionic character. Once the Si(100) surface is cooled below 40 K, the thermal motion of surface atoms is reduced and the surface silicon atoms form rows of dimers in a buckled and staggered manner, as shown in Fig. S1. Figure S1. The surfaces reconstruction of Si(100)-4x2 surface at cryogenic temperature. This surface structure is denoted Si(100)-4x2 surface, because the unit cell of the surface equals to a four-atom by two-atom cell. Under realistic thermal conditions (above room temperature), the dimers are in a fast dynamic equilibrium with a very shallow potential energy landscape. However, for the purposes of the computational description, we will consider the zwitterionic character of each dimer. Section 2. Comparison of different cluster models and basis sets Table S1: Comparison of predicted energies (kJ/mol, vs. reactant level) for selected structures at the B3LYP/LANL2DZ level of theory. Two different cluster models representing a hydroxylated Si surface site, (SiH3)3Si-OH and Si9H12-H2O, show very similar results and are indicative of the strongly localized character of the reactive site on the surface. Precursor Al(CH3)3 Hf[N(CH3)2]4 [CH3-C5H4]2Hf[CH3]2 Cu[acac]2 Ni[amd]2 Cluster model (SiH3)3Si-OH (Si9H13)-OH (SiH3)3Si-OH (Si9H13)-OH (SiH3)3Si-OH (Si9H13)-OH (SiH3)3Si-OH (Si9H13)-OH (SiH3)3Si-OH (Si9H13)-OH structure D -86.0 -86.0 -41.2 -41.8 -13.7 -10.9 -36.4 -44.2 -44.5 -46.5 E 0 -0.6 2.8 -7.8 -2 Kinetic barrier 55.0 53.6 5.5 0.0 26.0 43.7 17.8 24.5 -50.6 E -1.4 -5.5 17.7 6.7 -- structure E -199.0 -201.3 -154.8 -140.2 -126.9 -126.6 -31.4 -32.8 -83.1 -90.4 E -2.3 14.6 0.3 -1.4 -7.3 Table S2: Comparison of predicted energies (kJ/mol vs. reactants level) for selected structures during the ligandmediated adsorption of Al(CH3)3 using a Si12H15 (double dimer) surface model and a Si35H32 (four dimer) surface model. In both cases, the B3LYP/LANL2DZ level was used. The use of a larger cluster results in an increased stabilization but the observed trends in all the energies are consistent with those observed with smaller cluster models. AlMe3 Si35H32 (two-dimer cluster) Si15H16 (four-dimer cluster) structure A structure M structure H structure C -16.5 -274.4 -132.8 -452.0 -11.6 -242.3 -40.0 -380.3 E -4.9 -32.1 -92.7 -71.7 Table S3: Comparison of predicted energies (kJ/mol, vs. reactant level) for the structures corresponding to the simulated ligand-exchange reactions of metalorganic precursors on Si9H12-H2O model with B3LYP and LANL2DZ or 6311+G(d,p) basis sets. structure Kinetic structure E E E D barrier E 6311+G(d,p) -56.3 52.7 -178.0 Al(CH3)3 -29.7 0.9 -23.3 LANL2DZ -86.0 53.6 -201.3 6311+G(d,p) -38.1* 44.7 -47.9 Ti[O(C3H7)]4 -5.5 -8.3 -11.1 LANL2DZ -43.6 36.4 -59.0 6311+G(d,p) -7.9** 23.7 12.8 Cu[acac]2 -36.3 0.8 -45.6 LANL2DZ -44.2 24.5 -32.8 6311+G(d,p) -33.5 66.5 -62.5 Ni[amd]2 -13 -27.9 15.9 LANL2DZ -46.5 50.6 -90.4 * The optimization of this structure was performed with a lower convergence criteria to avoid complications with the multiple rotations of the iso-propoxyl groups. ** Only single point energy calculation result is provided for comparison. Precursor Basis set Table S4: Comparison of predicted energies (kJ/mol, vs. reactant level) for the structures corresponding to the simulated ligand-mediated adsorption reactions of Cu(acac)2 precursor on Si15H16 model using B3LYP and LANL2DZ or single-point using cc-pVTZ basis sets (from structures optimized at the LANL2DZ level). Although calculation with LANL2DZ basis set generally overestimates the energies, the observed trends for the reaction path are consistent for both LANL2DZ and cc-pVTZ basis sets. Cu(acac)2 structure A cc-pVTZ LANL2DZ -87.9 -70.6 -16.4 Difference transition state -89.7 -68.0 -21.7 structure M -166.7 -138.6 -28.1 transition state -76.0 -62.4 -13.6 Structure C -155.8 -137.5 -18.3 Section 3: Ethene elimination for AlEt3 precursor on Si(100) dimer. As shown in Figure S2, triethyl aluminum undergoes a facile C-H scission at the position. The low kinetic barrier (~5 kJ/mol) contrasts the one obtained for trimethyl aluminum (~200 kJ/mol), which does not feature hydrogen atoms. Figure S2. Ethene elimination pathway for a reaction of triethyl aluminum precursor and Si(100) surface investigated at a B3LYP/6311+G(d,p) level of theory. Section 4: Propene elimination for Ti[O(C3H7)]4 reaction. A simplified molecule where three of the iso-propoxy ligands were replaced with methoxy (CH 3O) ligands was employed; the remaining iso-propoxy group was used to investigate the elimination of both acetone and propene (Figure S3). Notice that propene elimination is more favorable thermodynamically than acetone elimination. Figure S3. Ligand decomposition possibilities of an iso-propoxy ligand (B3LYP/LANL2DZ). Section 5: Ligand elimination for Hf[C5H5]2[CH3]2 on OH-terminated Si dimer. Ligand elimination in Hf[C5H5]2[CH3]2 can start with either methyl or Cp group, as shown in Figure S4. Although the barriers for both pathways are comparable, methane elimination is predicted to lead to a more stable surface species. Figure S4. Ligand-exchange pathways for Hf[C5H5] 2[CH3] 2 (B3LYP/LANL2DZ). Section 6. Adsorption of Cu[acac]2 molecule. As summarized in Figure S5, adsorption of Cu[acac]2 on a bare Si surface occurs through the simultaneous attachment of both O and Cu. The second ligand is able to attach to the surface forming an additional Si-O bond with a rather large (yet feasible to oevrcome) energy barrier of 56 kJ/mol. Scission of a C-H bond from a methyl group is also predicted to be kinetically feasible (86 kJ/mol). It further stabilizes the structure (-220 kJ/mol). Notice that in this case the increase in stability is possible without forming a Si-C bond, because the ligand can stabilize the resulting CH2 moiety by forming a carbon-carbon double bond. Figure S5. Dissociative adsorption of Cu[acac] 2 molecule on the Si(100) surface (LANL2DZ). In order to avoid the complications presented by this mechanism and also to investigate the reactions of ligands attached to the transition metal center explicitly, in the manuscript the initial adsorption of the Cu[acac]2 molecule is considered onto a silicon surface dimer that has a single hydrogen atom attached to it (=Si-SiH= structure). The neighboring silicon dimer is unoccupied and is capable to provide reactive sites for further transformations of the ligands involved. The computational model cluster representing two neighboring surface silicon dimers with a single hydrogen atom attached to one of them is further used as a reactant in relative energy calculations presented in the text of the manuscript.