doc
advertisement

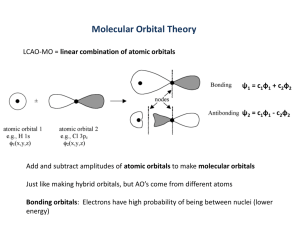
1 Molecular-Orbital Theory Introduction Orbitals in molecules are not necessarily localized on atoms or between atoms as suggested in the valence bond theory. Molecular orbitals can also be formed the LCAO where more than two atomic orbitals are used. (Technically, we can use all of the atomic orbitals in the LCAO.) Linear combinations of orbitals result in the interference of waves. Both constructive and destructive interference may result. Within a diatomic molecule, N atomic orbitals on atom 1 and N atomic orbitals on atom 2 result in 2N molecular orbitals. **In a practical sense, only those atomic orbitals that have similar energies and the appropriate symmetry can combine to form molecular orbitals.** These molecular orbitals are classified as bonding orbitals, antibonding orbitals or nonbonding orbitals. Types of Molecular Orbitals Bonding Orbitals For the linear combination of only two atomic orbitals, the bonding orbital is constructed from adding the wavefunctions together. MO AO A AO B Thus, the probability density of the bonding orbital can be written as MO AO A AO B 2 AO A AO B 2 2 2 A 2 B2 2AB The 2AB term is the constructive interference of the overlap of the atomic orbitals. - The interference results in an increase of electron density between the nuclei. - Thus, the nuclei have greater attraction to each other, via their mutual attraction to the increased electron density. 1s 1s 2px 2px 1s 2p 2 Aside: S AB d is called overlap integral. Antibonding Orbitals For the linear combination of only two atomic orbitals, the antibonding orbital is constructed from subtracting the wavefunctions together. MO AO A AO B Thus, the probability density of the bonding orbital can be written as MO AO A AO B 2 AO A AO B 2 2 2 A 2 B2 2AB The -2AB term is the destructive interference of the overlap of the atomic orbitals. - The interference results in a decrease of electron density between the nuclei and increase of electron density away from the nuclei. - Thus, the nuclei are drawn away from each other. 1s 2px 1s 1s 2px 2p The energy increase of the antibonding orbital is slightly higher than the energy decrease 1 E of the bonding orbital. 1 S Example: Let S 1 4 E 1 1 1 4 1 S 1 1 5 5 4 4 E 1 1 1 4 1 S 1 1 3 3 4 4 3 Energy Level Diagrams of Diatomic Molecular Orbitals Homonuclear Diatomic Molecules Hydrogen Bond order = ½ (# of bonding e- – # of antibonding e-) = number of bonds The bond order for the hydrogen molecule is one. Helium The bond order for the helium molecule is zero, i.e., He2 does not exist. 4 Lithium Dilithium exists! (Though not as crystal!) The bond order for the lithium molecule is one, i.e., 2 bonds – 1 antibond = 1 bond. 5 Beryllium Diberyllium does not exist. The bond order for the beryllium molecule is zero, - i.e., 2 bonds – 2 antibonds = 0 bond. 6 Molecular Orbital Diagrams for Second Row Diatomic Molecules B2 through N2 Notes: For B through N, 2s and 2p are close enough in energy for hybridization to change the order of the molecular orbital energies. B2 bond is bond?!? Also, B2 is paramagnetic. - Molecular electronic structure also follows Hund’s rules. C2 bonding is 2 bonds. C2 is diamagnetic. Note carbide ion, C22- has triple bond. N2 has triple bond. N2 is diamagnetic. Q: Which is more stable, N2+ or N2-? A: Antibond stronger than bond; therefore N2+ is predicted to be more stable. 7 O2 through Ne2 Notes: O2 bond is 1 and 1 bond. Also, O2 is paramagnetic. F2 bonding is bond. F2 is diamagnetic. Bond order of Ne2 is zero. Ne atoms are not bound. - Dimer does exist as van der Waals state. - Ne2+ can exist. Q: According to MO theory, Ne22+ should be stable ion. Why haven’t we heard of it until now? A: I don’t know. Hypothesis: Neon is even more electronegative than fluorine. 8 Heteronuclear Diatomic Molecules Energy of atomic orbitals is affected by effective nuclear charge. More electronegative atom will draw electron density to itself in a bonding orbital. Consider carbon monoxide as an example. C O C C O O C O Note: Most electropositive atom has most anti-bonding density. 9 Term Symbols of Molecular States Molecular Angular Momentum of Diatomic Molecules Orbital Angular Momentum The electron in a non-sigma bond experiences a cylindrically symmetric potential energy in contrast to an electron in an atomic non-s orbital that has a spherically symmetric potential energy. The electron in a non-sigma bond has an angular momentum, L. However, because the electric field from the nuclei and the other electrons is not spherically symmetric, the angular is not well characterized. However, for an electron in a non-sigma bond, the projection of the angular momentum upon the internuclear axis, ML, is well characterized. - To distinguish from the atomic ML, the molecular ML is given a new symbol, . Thus, the electronic state of a diatomic molecule is partially characterized by . symbol 0 1 2 3 Each electron contributes to the total orbital angular momentum thus 1 2 Each state is doubly degenerate except the state that has one state. In other words, E E A non-sigma bond electron has two directions to precess about the internuclear axis. Spin Angular Momentum The spin precesses about the internuclear axis since the magnetic moment of the electron tries to align itself with the magnetic field created by the “nuclei orbiting about the electron”. - This phenomenon could be referred to as molecular spin-orbit coupling. The projection of the spin, S, on the internuclear axis is MS. - To distinguish from the atomic MS, the molecular MS is given a new symbol, . - Note source of confusion: can refer to a specific orbital angular momentum state or it can refer to the general total spin angular momentum. Each electron contributes to the total spin angular momentum thus 1 2 Multiplicity = 2 + 1 The rules for multiplicity are the same as for atomic structure. - filled orbital multiplicity = 1 singlet 0 - half-filled orbital multiplicity = 2 doublet 1 2 - two half-filled orbitals 1 multiplicity = 3 triplet 10 Introduction to Symmetry Parity g – gerade symmetric for inversion through center u – ungerade antisymmetric for inversion through center Reflection + - reflection through plane containing internuclear axis is symmetric – - reflection through plane containing internuclear axis is antisymmetric Consider the symmetries of the sigma and pi bonding and antibonding orbitals. Parity Reflection g + u – u + g – When two or more electrons are in a molecular state, the total symmetry of the state is determined by multiplying the symmetries of the electrons together. Multiplication rules for of parity and reflection symmetries Parity gg g gu ug u u u g Reflection Writing Term Symbols Information available in a molecular term symbol multiplicity orbital angular momentum 2 g reflection symmetry parity 11 Writing an electron configuration as a term symbol. Use B2 as an example. The electron configuration for B2 can be written as (1s2)(1s*2)(2s2)(2s*2)(2p2) As in the atomic case, closed subshells do not contribute to the total angular momentum. Two cases for electrons in the 2p orbitals. each electron is in different orbitals state 1 2 1 1 0 electron spins can be parallel or antiparallel 1 2 1 2 1 2 1 or 1 2 1 2 1 2 0 thus, possible terms are 3 g or 1 g both electrons in the same orbital state 1 2 1 1 2 electron spins can be antiparallel only (can’t violate Pauli exclusion principle) 1 2 1 2 1 2 0 thus, possible terms are 1 g Using Hund’s rules as with the atomic term symbols, the term with the highest multiplicity is the lowest energy. Thus the ground state term of B2 is 3 g Use C2 as another example. The ground state electron configuration for C2 can be written as (1s2)(1s*2)(2s2)(2s*2)(2p4). Thus the term for the ground state is 1 g . However, consider excited state 3 u . What is the electronic configuration for lowest possible excited state? How can we assign the molecular orbitals for the various electrons? The state implies that = 0. At this point, let us consider that a single electron has been excited to a * orbital. 0 1 2 0 1 1 2 1 Note that the above could be true if the electron is excited to a higher orbital as well. multiplicity = 3 implies that 1 1 2 1 2 12 The one electron remaining in the orbital has an ungerade parity and negative reflection symmetry. Thus, the electron in the unknown orbital must be gerade parity and negative reflection symmetry. u ? u ? g ? ? Since the * orbital has gerade parity and negative reflection symmetry, it is possible that the 3 u implies the electron configuration (1s2)(1s*2)(2s2)(2s*2)(2p3)(2p*1). However, ambiguity exists in the term symbol. The electron configuration, (1s2)(1s*2)(2s2)(2s*2)(2p3)(3p*1), would also result in a term of 3 u . To unambiguously assign electronic structure, more information is needed. The term symbols of molecular states are very convenient when studying electronic spectroscopy. The selection rules of electronic spectroscopy are dependent on the symmetry changes of the states. Thus having the term symbols will allow us to quickly identify between which states transitions can occur. 13 Molecular Orbitals of Polyatomic Molecules The molecular orbitals of polyatomic molecules can be constructed from adding and subtracting appropriate combinations of atomic orbitals. The atomic orbitals must have a proper spatial symmetry in order for a net bond overlap to occur. A Little Trick – Symmetry Adapted LCAO Consider the construction of molecular orbitals for a polyatomic molecule like linear H3. It is more difficult to consider linear combinations of three orbitals rather than two orbitals. Therefore, we will be clever and consider only combinations of two orbitals at a time. H3 – Linear Triatomic Hydrogen For H3, we will create bonding and antibonding orbitals from the atomic orbitals of hydrogens 1 and 3. Then we will combine the molecular orbitals of hydrogens 1 and 3 with the atomic orbital of hydrogen 2 to make a complete set of molecular orbitals. First add and subtract the 1s orbitals of hydrogens 1 and 3. + 1s 1s 1s 1s 1s - 1s Note that since hydrogens 1 and 3 are far apart, very little overlap occurs. An energy level diagram demonstrates that the energy changes at this point are minimal. Now let us take each molecular orbital above and add the 1s orbital of hydrogen 2 to each. 14 Orbital 1 Note that orbital 1 bonds all three hydrogen nuclei. No Interaction There is no interaction between the 1s* and 1s since the amount of positive overlap equal the amount of negative overlap. Thus no new orbital is formed. Now take the molecular orbitals from hydrogens 1 and 3 and subtract the 1s orbital of hydrogen 2 from each. Orbital 3 Note that orbital 3 is antibonding between all three hydrogen nuclei. No Interaction Again the interaction between the 1s* and 1s is zero. 15 If we have formed two molecular orbitals from three atomic orbitals so far, where is the third orbital? The third orbital has the shape of the 1s* orbital. Orbital 2 We note that orbital 2 does not draw nuclei together as a bonding orbital would do nor does it draw nuclei apart as an antibonding orbital would do. Thus, orbital 2 is named a nonbonding orbital. The energy level diagram for these molecular orbitals is Thus linear H3 has a bonding orbital (orbital 1), a nonbonding orbital (orbital 2) and an antibonding orbital (orbital 3). H2O – Water To find the molecular orbitals for water we will take a similar tack as we did with H3. First we will construct , and * orbitals from the hydrogen 1s orbitals. Now let us consider the interaction of these orbitals with the five orbitals of oxygen, 1s, 2s, 2px, 2py and 2pz. First we can consider that the 1s orbital of oxygen is much lower in energy than hydrogen 1s orbitals. Therefore, as a first approximation, we will consider that there is no interaction with the 1s orbital of oxygen. The 1s orbital of oxygen is a nonbonding orbital or a perhaps better described as a nonbonding core orbital. 16 Next, consider the interaction with the 2s orbital. The energy of the 2s oxygen orbital is still below the energy of the 1s hydrogen orbital; but, it is close enough for an interaction to occur. The shapes of the orbitals are similar to the H3 orbitals but the water orbitals are bent. No Interaction The 1s* orbital may be a nonbonding orbital or it may interact with another orbital. Now consider the interaction with 2p orbitals. First we need to define a coordinate system. Z Y H H O X Now consider the interaction between the hydrogen orbitals and the 2py orbital. Looking down from z direction. 17 Note that any constructive interference that occurs is balanced by the destructive interference that occurs. Thus, the effective interaction is zero. The lack of interaction occurs whether we consider the hydrogen orbitals in the 1s or the 1s* configuration. Therefore, the 2py orbital is a nonbonding orbital. Now consider the interaction with the 2pz orbital. Let us add the 2pz orbital to the hydrogen molecular orbitals. No Interaction We still don’t know if the 1s* orbital will be a nonbonding orbital or whether it will interact with another orbital. Also, note that we could subtract the 2pz orbital from the 1s orbital to yield a strictly antibonding orbital. Finally, consider the interaction with the 2px orbital. No Interaction Note that we could subtract the 2px orbital to yield a strictly antibonding orbital. Finally we answer the question about the 1s* orbital. It will interact with the 2px orbital to form a bonding and antibonding orbital. 18 Hückel Model Previous discussion has been a simplified view of MO theory. Procedures exist to take all of the atomic orbitals and mix them together to form molecular orbitals. Examining the procedure is beyond what we would like to consider here. However, we get a taste of the procedure by examining what is known as the Hückel model. The Hückel model is applicable to the pi system of conjugated molecules. 1. Construct wavefunction from a sum of p orbitals perpendicular to the molecular plane. c1p1 c2p2 c3p3 2. Assume that all overlap integrals are zero. S pi p j ij 0 3. Assume H11 H22 H33 Hii pi H pi 4. Assume that H12 H23 H34 for adjacent p orbitals Hij pi H p j 5. Assume that H13 H14 H24 0 for nonadjacent p orbitals 6. Then construct a secular determinant. Consider cyclobutadiene as an example. 0 c1 E E 0 c2 0 0 E c3 0 E c4 E 0 0 E 0 0 E 0 E 7. Make solution of secular determinant easier by performing the substitution, E x x 1 0 1 1 x 1 0 0 0 1 x 1 1 0 1 x 19 8. Find the solutions of the secular determinant. x 4 4x 2 0 x 0,0, 2, 2 E , , 2, 2 9. Find the eigenvectors of the secular determinant to find the molecular orbitals For E 2 v 1 2 1 2 1 2 1 2 For E v 1 For E v 0 1 For E 2 v 1 2 1 2 1 2 1 2 2 0 1 2 0 2 0 1 2 1 1 1 1 p1 p2 p3 p4 2 2 2 2 1 1 p p 2 1 2 3 1 1 p2 p 2 2 4 1 1 1 1 p1 p2 p3 p4 2 2 2 2