Vibrations
advertisement

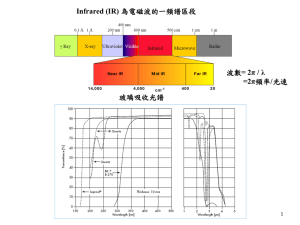
Infra-Red (IR) Spectroscopy or Vibrational spectroscopy Applied Chemistry Course: CHY101 Electromagnetic Radiation Frequency () Wavelength () Radio-waves Region Microwaves Region Infra-red Region Visible Region Ultra-violet Region X-ray Region -ray Region Frequency (HZ) 106 - 1010 1010 - 1012 1012 - 1014 1014 - 1015 1015 - 1016 1016 - 1018 1018- 1020 Wavelength 10m – 1 cm 1 cm – 100µm 100µm – 1µm 700 – 400 nm 400-10 nm 10nm – 100pm 100pm – 1 pm NMR, ESR Rotational Spectroscopy Vibrational spectroscopy Electronic Spectroscopy Electronic Spec. 0.001 – 10 J/mole Order of some 100 J/mole Some 104 J/mole Some 100 kJ/mole Some 100s kJ/mole 107- 109 J/mole 109- 1011 J/mole Energy UHF TV, cellular, telephones. (300 MHz and 3 GHz) FM Radio, VHF TV. AM Radio Microwave ovens, Police radar, satellite stations-- (3 to 30 GHz) Table lamp, Tube light (400 nm -800 nm or 400–790 THz) Sun Lamp X-ray Electromagnetic Radiation The interaction of radiation with matter http://hyperphysics.phy-astr.gsu.edu/hbase/mod3.html http://www1.lsbu.ac.uk/water/vibrat.html Hook’s Law Hook’s Law, F=-kx F is the restoring force x is the displacement from equilibrium k is the force constant The restoring force proportional to the displacement of particle from its equilibrium position and the force constant k of the spring. Vibrational spectra: Harmonic oscillator model The Simple Harmonic Oscillator Model: H2 molecule H1 H2’ H2” E2 In this case, energy curve is parabolic, E= E1 H1 H2” H2’ req 1 k (r – req)2 2 Harmonic Oscillator Model The Simple Harmonic Oscillator Model: H2 molecule H1 The compression & extension of a bond (like a spring) obeys Hooke’s Law, H2’ H2” E2 E1 H1 H2” H2’ req f is the restoring force k is the force constant In this case, energy curve is parabolic, E= 1 k (r – req)2 2 Anharmonic Oscillator Model Where a is a constant for a particular molecule and Deq is the dissociation energy. The Morse potential (blue) and harmonic oscillator potential (green). The potential at infinite internuclear distance is the dissociation energy for pure vibrational spectra. Anharmonic oscillator model The actual potential energy of an anharmonic oscillator fits the parabolic function fairly well only near the equilibrium internuclear distance. The Morse potential function more closely resembles the potential energy of an anharmonic oscillator model. Where a is a constant for a particular molecule and Deq is the dissociation energy. Deq req Fig. The energy diagram of molecule’s vibrational model showing an (a) ideal diatomic or (b) anharmonic diatomic oscillator. http://www1.lsbu.ac.uk/water/vibrat.html Vibrational spectra: Harmonic oscillator model Oscillation Frequency, k is the force constant = reduced mass, Vibrational frequency only dependent on the mass of the system and the force constant . From Schrodinger equation, Vibrational energies for simple harmonic oscillator, Vibrational spectra: Harmonic oscillator model At Zero-point energy The implication is that the diatomic molecule (and, indeed, any molecule) can never have zero vibrational energy; the atoms can never be completely at rest relative to each other. The simple selection rule for the harmonic oscillator undergoing vibrational changes The allowed vibrational energy levels and transitions between them for a diatomic molecule undergoing simple harmonic motion. Anharmonic oscillator model The Morse potential function more closely resembles the potential energy of an anharmonic oscillator model. Energy in a particular vibrational energy level of an anharmonic oscillator is found to be (from Schrodinger equation), Deq req Where xe is the corresponding anharmonicity constant which, for bond stretching vibrations, is always small and positive (~ +0.01), so that the vibrational levels are crowd more closely together with increasing vibrational level v. Fig. The energy diagram of molecule’s vibrational model showing an (a) ideal diatomic or (b) anharmonic diatomic oscillator. Vibrational spectra: Anharmonic oscillator model Vibrational energies for simple harmonic oscillators, ……… A ……… B Vibrational energies for anharmonic oscillators, Vibrational spectra: Harmonic oscillator model Oscillation Frequency, k is the force constant = reduced mass, The vibrational frequency is increasing with: increasing force constant k ( = increasing bond strength) decreasing atomic mass • Example: k cc > k c=c > k c-c Infrared Spectroscopy POSITION REDUCED MASS LIGHT ATOMS HIGH FREQUENCY BOND STRENGTH (STIFFNESS) STRONG BONDS HIGH FREQUENCY The vibrational frequency is increasing with: • increasing force constant k (= increasing bond strength) • decreasing atomic mass Different types of Energy Connecting macroscopic thermodynamics to a molecular understanding requires that we understand how energy is distributed on a molecular level. ATOMS: The electrons: Electronic energy. Increase the energy of one (or more) electrons in the atom. Nuclear motion: Translational energy. The atom can move around (translate) in space. MOLECULES: The electrons: Electronic energy. Increase the energy of one (or more) electrons in the molecule. Nuclear motion: Translational energy. The entire molecule can translate in space. Vibrational energy. The nuclei can move relative to one another. Rotational energy. The entire molecule can rotate in space. N2 CO2 Molecular vibrations How many vibrations are possible (= fundamental vibrations)? A molecule has as many degrees of freedom as the total degree of freedom of its individual atoms. Each atom has three degrees of freedom (corresponding to the Cartesian coordinates), thus in an N-atom molecule there will be 3N degree of freedom. In molecules, movements of the atoms are constrained by interactions through chemical bonds. Molecular vibrations Molecular vibrations Molecular vibrations Translation - the movement of the entire molecule while the positions of the atoms relative to each other remain fixed: 3 degrees of translational freedom. Rotational transitions – interatomic distances remain constant but the entire molecule rotates with respect to three mutually perpendicular axes: 3 rotational freedom (nonlinear), 2 rotational freedom (linear). Fundamental Vibrations Vibrations – relative positions of the atoms change while the average position and orientation of the molecule remain fixed. Molecular vibrations Stretching and Bending Types of Molecular Vibrations: Vibrations fall into the basic categories of stretching and bending. Stretching vibration involves a continuous change in the inter-atomic distance along the axis of the bond between two atoms. Bending vibrations are characterized by a change in the angle between two bonds and are of four types: scissoring, rocking, wagging, and twisting. Stretching Vibration Bending Vibration H2O: Stretching and Bending Vibrations Bending is easier than stretching -- happens at lower energy (lower wavenumber) Bond Order is reflected in ordering -- triple > double > single (energy) with single bonds easier than double easier than triple bonds Heavier atoms move slower than lighter ones Examples of Vibrational Modes Example 1: Consider Water Examples of Vibrational Modes Example 1: Consider Water Vibrations of water Gas H216O 3756 cm-1 v1, cm-1 3657.1 v2, cm-1 1594.7 3657 cm-1 v3, cm-1 3755.9 1595 cm-1 Examples of Vibrational Modes Example 2: Consider Carbon Dioxide Examples of Vibrational Modes Example 3: The Methylene Group It is important to note that there are many different kinds of bends, but due to the limits of a 2-dimensional surface it is not possible to show the other ones. A linear molecule will have another bend in a different plane that is degenerate or has the same energy. This accounts for the extra vibrational mode. Selection Rules A molecule will absorb infrared radiation if the change in vibrational states is associated with a change in the dipole moment () of the molecule. Vibrations which do not change the dipole moment are Infrared Inactive (homonuclear diatomics). Homonuclear diatomic molecules (O2, N2, H2, Cl2) – IR Inactive Heteronuclear diatomic molecules (HF, HCl) – IR Active Dipole moment is greater when electronegativity difference between the atoms in a bond is greater. Some electronegativity values are: H 2.2; C 2.55; N 3.04; O 3.44; F 3.98; P 2.19; S 2.58; Cl 3.16 Infrared Spectrum of Carbon Dioxide (CO2) Stretching Bending Asymmetric Stretching Vibration of the CO2 Fig. The asymmetric stretching vibration of the CO2 molecule, showing the fluctuation in the dipole moment. The asymmetric stretch is infrared active because there is a change in the molecular dipole moment during this vibration. Symmetric Stretching Vibration of the CO2 To be "active" means that absorption of a photon to excite the vibration is allowed by the rules of quantum mechanics. [Aside: the infrared "selection rule" states that for a particular vibrational mode to be observed (active) in the infrared spectrum, the mode must involve a change in the dipole moment of the molecule.] The symmetric stretch is not infrared active, and so this vibration is not observed in the infrared spectrum of CO2. Bending motion of the CO2 Fig. The bending motion of the carbon dioxide molecule and its associated dipole fluctuation. Infrared Spectroscopy POSITION STRENGTH REDUCED MASS LIGHT ATOMS HIGH FREQUENCY BOND STRENGTH (STIFFNESS) STRONG BONDS HIGH FREQUENCY CHANGE IN ‘POLARITY’ STRONGLY POLAR BONDS INTENSE BANDS The vibrational frequency is increasing with: • increasing force constant k (= increasing bond strength) • decreasing atomic mass Calculated IR bands for CH2 in formaldehyde (CH2O) Infrared Spectroscopy 4000-3000 cm-1 3000-2000 cm-1 2000-1500 cm-1 1500-1000 cm-1 1000- below cm-1 O-H N-H C-H C C C N C=C C=O C-O C-F C-Cl C-C deformations Bending/other stretching – Fingerprint region increasing energy increasing frequency The vibrational frequency is increasing with: • increasing force constant k = increasing bond strength • decreasing atomic mass • Example: k c c > k c=c > k c-c Absorption Regions Infrared Spectroscopy for Structure Determination Divide the spectrum in to two regions: 4000 cm-1 1600 cm-1 most of the stretching bands, specific functional groups (specific atom pairs). This is the “functional group” region. 1600 cm-1 400 cm-1 many bands of mixed origin. Some prominent bands are reliable. This is the “fingerprint” region. Use for comparison with literature spectra. Alcohols O–H stretch, hydrogen bonded 3500-3200 cm-1 C–O stretch 1260-1050 cm-1 (s) C-C stretches C–O stretch in same region as C–C but much more intense. O-H Infrared Spectrum of EtOH (Ethanol) Alcohols C–O–H stretch 3600 cm-1 in dilute solution phenethanol Typically H-bonding and at lower frequency ~3400 cm-1 Position is sensitive to subs. pattern 1-phenylethanol RCH2–OH 1050 cm-1 R2CH–OH 1110 cm-1 R3C–OH 1175 cm-1 Why O-H peak is broad? 2-piperidinylethanol Ketones C=O stretch: aliphatic ketones 1715 cm-1 alpha, beta-unsaturated ketones 1685-1666 cm-1 Carbonyl Compounds All carbonyl compounds absorb in the region 1760-1665 cm-1 due to the stretching vibration of the C=O bond. Aldehydes H–C=O stretch 2830-2695 cm-1 C=O stretch: aliphatic aldehydes 1740-1720 cm-1 alpha, beta-unsaturated aldehydes 1710-1685 cm-1 The carbonyl stretch C=O of saturated aliphatic aldehydes appears from 1740-1720 cm-1. As in ketones, if the carbons adjacent to the aldehyde group are unsaturated, this vibration is shifted to lower wavenumbers, 1710-1685 cm-1. Aldehydes Note: The O=C–H stretches in both aldehydes in the region 2830-2695 cm-1, especially the shoulder peak at 2725 cm-1 in butyraldehyde and 2745 cm-1 in benzaldehyde. Carboxylic Acids Carboxylic acids show a strong, wide band for the O–H stretch. Unlike the O–H stretch band observed in alcohols, the carboxylic acid O–H stretch appears as a very broad band in the region 3300-2500 cm-1, centered at about 3000 cm-1. O–H stretch from 3300-2500 cm-1 C=O stretch from 1760-1690 cm-1 C–O stretch from 1320-1210 cm-1 O–H bend from 1440-1395 and 950-910 cm-1 Esters The carbonyl stretch C=O of aliphatic esters appears from 1750-1735 cm-1; that of α, β-unsaturated esters appears from 1730-1715 cm-1. C=O stretch aliphatic from 1750-1735 cm-1 α, β-unsaturated from 1730-1715 cm-1 C–O stretch from 1300-1000 cm-1 Esters Note: The C=O stretch of ethyl acetate (1752) is at a higher wavelength than that of the α, β-unsaturated ester ethyl benzoate (1726). Also note the C–O stretches in the region 1300-1000 cm-1. Amines The N–H stretches of amines are in the region 3300-3000 cm-1. These bands are weaker and sharper than those of the alcohol O–H stretches which appear in the same region. In primary amines (RNH2), there are two bands in this region, the asymmetrical N–H stretch and the symmetrical N–H stretch. Secondary amines (R2NH) show only a single weak band in the 3300-3000 cm-1 region, since they have only one N–H bond. Tertiary amines (R3N) do not show any band in this region since they do not have an N–H bond. N–H stretch 3400-3250 cm-1 1° amine: two bands from 3400-3300 and 3330-3250 cm-1 2° amine: one band from 3350-3310 cm-1 3° amine: no bands in this region N–H bend (primary amines only) from 1650-1580 cm-1 C–N stretch (aromatic amines) from 1335-1250 cm-1 C–N stretch (aliphatic amines) from 1250–1020 cm-1 N–H wag (primary and secondary amines only) from 910-665 cm-1 Amines The spectrum of aniline is shown below. This primary amine shows two N–H stretches (3442, 3360); note the shoulder band, which is an overtone of the N–H bending vibration. The C–N stretch appears at 1281 rather than at lower wavenumbers because aniline is an aromatic compound. Also note the N–H bend at 1619. Amines The spectrum of diethylamine is below. Note that this secondary amine shows only one N–H stretch (3288). The C–N stretch is at 1143, in the range for non-aromatic amines (1250-1020). Diethylamine also shows an N–H wag (733). Amines Triethylamine is a tertiary amine and does not have an N–H stretch, nor an N–H wag. The C–N stretch is at 1214 cm-1 (non-aromatic). Amines R-NH2 3500, 3300 cm-1 doublet, frequently NH stretch (without, with H-bonding effect) 1600 cm-1 NH2 scissoring - broad 700-900 cm-1 NH2 wagging - broad, strong 1080 cm-1 C–N str. --weak for alkyl 1300 cm-1 Ar–N str. strong R–NH–R 3400 cm-1 singlet str. NH stretching Weak C–N 1125 cm-1 R–NR–R No IR band over 3000 cm-1, NO NH bond. adjacent CH2 will shift to 2800 cm-1. A tert-amine salt NH strong at 2500 cm-1 Alkanes C–H stretch from 3000–2850 cm-1 C–H bend or scissoring from 1470-1450 cm-1 C–H rock, methyl from 1370-1350 cm-1 C–H rock, methyl, seen only in long chain alkanes, from 725-720 cm-1 Alkenes C=C stretch from 1680-1640 cm-1 =C–H stretch from 3100-3000 cm-1 =C–H bend from 1000-650 cm-1 Alkynes –C≡C– stretch from 2260-2100 cm-1 –C≡C–H: C–H stretch from 3330-3270 cm-1 –C≡C–H: C–H bend from 700-610 cm-1 Alkyl Halides C–H wag (-CH2X) from 1300-1150 cm-1 C–X stretches (general) from 850-515 cm-1 C–Cl stretch 850-550 cm-1 C–Br stretch 690-515 cm-1 Alkyl Halides Aromatics C–H stretch from 3100-3000 cm-1 overtones, weak, from 2000-1665 cm-1 C–C stretch (in-ring) from 1600-1585 cm-1 C–C stretch (in-ring) from 1500-1400 cm-1 C–H "oop" from 900-675 cm-1 Out-of-plane CH bending Nitro Groups The N–O stretching vibrations in nitroalkanes occur near 1550 cm-1 (asymmetrical) and 1365 cm-1 (symmetrical), the band at 1550 cm-1 being the stronger of the two. If the nitro group is attached to an aromatic ring, the N–O stretching bands shift to down to slightly lower wavenumbers: 1550-1475 cm-1 and 1360-1290 cm-1. N–O asymmetric stretch from 1550-1475 cm-1 N–O symmetric stretch from 1360-1290 cm-1 Nitro Groups Compare the spectra of nitromethane and m-nitrotoluene: In nitromethane, the N–O stretches are at 1573 and 1383, while in nitrotoluene, they are a little more to the right, at 1537 and 1358. Nitro Groups + N O Analogous to Carboxylate ion. Strong bands Aromatic nitro: 1520, 1350 cm-1 O R + N O O Aliphatic nitro: 1550, 1370 cm-1 Esters and Lactones O 1735 cm-1 O R 1300-1100 intense, often doublet Determining benzene ring substitution patterns from IR spectra IR spectra provide valuable information about local configurations of atoms in molecules. The three possible isomers are easy to differentiate by IR. It can be used to identify the relative positions of the substituents on a disubstituted benzene ring. Examples are: