Bio321_2010 slides
advertisement

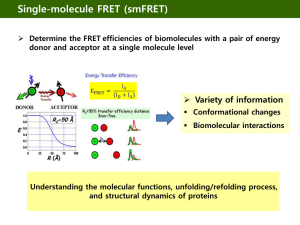
Microscopy is about a combination of resolution (seeing smaller and smaller things), and contrast (seeing what you want to see). Both aspects have recently seen great advancements, with the advent of single molecule studies (in dilute solutions) and super-resolution, beating the Abbe-limit. Here, we will treat both aspects, starting with contrast agents that are either added or that occur naturally. Contrast - Polarization, birefringence - Fluorescence lifetime - Fluorescence transfer Resolution/Contrast - Two Photon Microscopy - Single plane illumination Resolution - Stimulated Emission Depletion - Photo-activated localization - Structured Illumination -Total internal reflectance - Scanning near field Polarization I property of light waves, describes orientation of oscillations http://www.ifsc.usp.br/~lavfis/BancoApostilasImagens/ApEfFotoelastico/photoelasticity.pdf Polarization microscopy Polarization II http://www.ifsc.usp.br/~lavfis/BancoApostilasImagens/ApEfFotoelastico/photoelasticity.pdf Birefringence ellipticity f(d) = (n|| – n ) d/l Results in conoscopic figures Problem! cholesteric liquid crystals nematic liquid crystals Twisted liquid crystal cell = polarization switch = light switch - can be used as a filter (Kerr effect) Used in flat screens (TV, notebooks, beamers…) , TFT LCD = thin film transistor liquid crystal display Birefringence image is INDEPENDENT of orientation of optical axis Microtubule aster image White: 0.3nm retardance movie Fluorescence Fluorescence principle Vibrational states Light basically interacts with the electrons in a given material. The photons making up the light in Quantum mechanics can be absorbed, if there is an electron state at the energy corresponding to the absorbed energy of the photon. Once a photon is absorbed and the electrons are excited, they can either relax via collisions and vibrations or by emitting another photon. In case the electron relaxes before emitting another photon, there is fluorescence. Natural proteins for fluorescence studies Green fluorescent protein (GFP) Fluorescence lifetime microscopy (FLIM) Lifetime image A. GFP-tagged protein B. YFP- tagged protein C & D. Both GFP and YFP tagged proteins. The colour bars show the calibration of fluorescence lifetime from approx 2.1ns (red) to 3.0ns (dark blue). Measuring viscosities using FLIM Measuring temperatures using FLIM Forster resonance energy transfer (FRET) Overlap of emission and absorption spectra of flouorophores can be used to obtain information on their distance FRET measures distance changes in the nanometer scale, a relevant length scale for many biomolecules. Histone phosphorylation (specific for serine 28 on histone 3) in living HeLa cells undergoing cell division. Red signifies high FRET (and high phosphorylation levels), blue signifies low FRET (and low phosphorylation levels), and green is intermediate. The reporter displays a rapid increase in FRET 5-15 minutes after breakdown of the nuclear envelope. Functional studies using FRET on single molecules in real time Time record of folding and unfolding of an RNA molecule hairpin ribozyme. We attach the donor (green) and acceptor (red) dyes to the RNA so that the folded state has high FRET and the unfolded state has low FRET. We can see this beautiful two-state fluctuations in FRET values as a function of time Two photon fluorescence microscopy Principle of 2 photon fluorescence Radiationless decay fluoro weakest absorption Best penetration near 1000nm 2 NIR photons are absorbed simultaneously Typical two photon fluorescence setup High intensity required ! 2 hv excit Fluorescence detection Selective plane illumination microscopy (SPIM) Scanning SPIM (DLSM) Allows for faster scanning and induces less photons to the sample, since only a single line is illuminated. Long-time imaging becomes possible. Movie of Zebrafish embryo nuclei Stimulated emission depletion (STED) microscopy Typical setup Photo-activated localization microscopy (PALM) The principle of PALM The principle of PALM needs a Photo-switchable probe Imaging laser (657 nm) Activator Cy3 Reporter Cy5 Activation laser pulses Cy5 fluorescence 0 6000 photons Activation Cy3 Cy5 Cy3 5 10 Time (s) Cy5 Activation laser (532 nm) 15 20 5 μm B-SC-1 cell, Microtubules stained with anti-β tubulin Cy3 / Alexa 647 secondary antibody 5 μm Bates et al, Science 317, 1749 – 1753 (2007) 500 nm 5 μm Standing wave illumination microscopy (SWIM) Also beats the Abbe limit Total internal reflection (fluorescence) microscopy TIRM z Fluorescence intensity I(z) ~ I0 Evanescent light wave I0, decays exponentially with distance z from surface I0 ~ exp(-az) z(t) ~ - ln I(t), fluctuating position Distribution p(z) ~ exp (-F(z)/kT) Typical potential energy curves of a negatively charged polystyrene sphere(R=5 µm) close to an equally charged glass surface as a function of the separation distance between the glass and the particle surface. For large particle-surface separations the interaction potential is dominated by gravity which can be seen in the linear behavior of the potential curve in this regime, whereas at small separations the repulsive Coulomb interaction dominates. The potential curves are plotted for particles with different weights Optical near field microscope (SNOM) D<l “near field “ Recap • Microscopy is all about resolution AND contrast. • Birefringence gives information on molecular properties. • Fluorescence lifetime and transfer can be used as contrast agents. • Contrast (and resolution) enhancement can be obtained by two-photon excitation and single plane illumination. • Resolution increase is possible by using structured illumination and ingeneous fluorescence excitation.