Aijun_H2 desorption from MgH2
advertisement

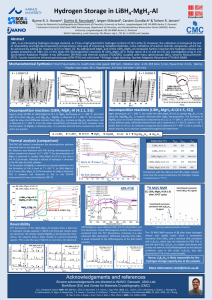
Ab intio Studies of Hydrogen Desorption from Low-Index Magnesium Hydride Surface Aijun Dua,b, Sean C. Smitha,b, X.D.Yaob,c and G.Q.Lub aCentre bARC for Computational Molecular Science, Chemistry Building, The University of Queensland, Qld 4072 Centre for Functional Nanomaterial, School of Engineering, The University of Queensland, Qld 4072 cSchool of Engineering, James Cook University, Townsville, Qld 4811 Results & Discussion (continued) Introduction Magnesium hydride is one of the most promising candidates in the automotive industry due to its very high capacity (7.6 %), light weight and low cost. Unfortunately, the practical application is primarily limited by the slow kinetics and the high operating temperature (> 573K). Experimentally, great efforts have been made to improve the H2 absorption and desorption kinetics by mechanically milling MgH2 [1] and adding transition metals [2]. These have been shown to work quite well to obtain the fast hydrogenation of Magnesium as reported by several groups [3-4]. However, in most case, it still requires at least 250oC to liberate hydrogen from magnesium hydride. In spite of many experimental studies above, little is known about the intrinsic physical and chemical properties of MgH2 system. Theoretically, ab initio DFT calculations have shown considerable predictive power for catalysis and could provide much insight on designing alloy catalyst [5]. To date, There have been some theoretical calculations to study the Mg-H bonding properties, but most of them are mainly limited in either less accurate cluster calculation or LDA only calculations[6-7], which have the strong overbinding. To my knowledge, very few calculations are performed to study hydrogen desporption from Magnesium hydride surface except for a cluster calculation [7]. In order to improve the hydrogen desorption performance, it is highly desired to understand the fundamental Mg-H bonding properties and activated barrier starting from a realistic Magnesium hydride surface, in particular from microscopic viewpoint. In the present study, the low-index Magnesium hydride surfaces, MgH2(001) and MgH2(110) were first studied with ab intio Density Functional Theory (DFT) calculation. Then the activated barriers for desorption of H2 from both surfaces are investigated utilizing the Nudged Elastic Band (NEB) method. (a) (b) (c) (d) Computational Methodology Ab initio PAW Method: The calculations were performed using the plane-wave basis VASP code [8] implementing the generalized gradient approximation (GGA) of PBE exchange correlation functional. An all-electron description, the projector augmented wave method (PAW) [9] is used to describe the ectronic-ion-core interaction. Within the PAW method, the lattice constant of bulk MgH2 is calculated to be 4.510 Å. and only 0.2 % in relative error compared the experimental value. The cutoff energy for plane waves is chosen up to of 400 eV for the accurate energy calculation. A (2×2) surface unit cell and a (4×4×1) Monkhost-Pack grid for the k-point sampling of the first Brillouin-zone are used in describing MgH2(001) surface. For the MgH2(110) surface, we chose a big supercell with 144 atoms included and only gamma point calculation is performed. The vacuum space is at least 15 Å, which is enough to guarantee a sufficient separation between periodic images. Nudged Elastic Band Method: To determine dissociation barriers and Minimum Energy Paths (MEP), the NEB method was used [10-11]. This method involves optimizing a chain of images that connect the reactant and product state. Each image is only allowed to move into the direction perpendicular to the hyper-tangent. Hence the energy is minimized in all directions except for the direction of the reaction path. A damped molecular dynamics was used to relax ions until the force in each image are less than 0.02 eV/Å. Figure 2 The projection of total valence electron charge density on (a) MgH2(001) and (c) MgH2(110) surface. The three dimensional iso-surface with a value of 0.01 electron for (a) MgH2(001) and (b) MgH2(110) surfaces, respectively. 2.5 3.0 Relative Energy (eV) Results & Discussion Relative Energy (eV) 2.0 2.5 2.0 1.5 1.0 0.5 1.5 1.0 0.5 0.0 0.0 0 2 4 6 8 10 12 Image Number Index 0 2 4 6 Image Number 8 10 12 Figure 3 The energy profiles for the desorption of two H atoms from (a) MgH2(001) and (b) MgH2(110) surface. Conclusions The low index Magnesium hydride surfaces, MgH2(001) and MgH2(110) were first studied with ab intio Density Functional Theory (DFT) calculation. Then the barriers for desorption of H2 from both surfaces are investigated utilizing the Nudged Elastic Band (NEB) method. We found that MgH2(110) surface is more stable than MgH2(001) surface, which is in good agreement with the experimental observation. The activated barrier for hydrogen desorption from the magnesium hydride surface is considerable due to its thermodynamic stability. It is about 0.45 eV higher for MgH2(001) surface than that on MgH2(110) surface. Our results compare favorably to the only existing cluster calculation in relation to the experimental results. In addition, we could conclude that the lower iconicity of hydrogen shall contribute to weak binding of hydrogen atoms, i.e., the higher performance of hydrogenation and dehydrogenation. Recently, some experimental studies have also shown d-electron transition metals, such as Nb and V may be acceptable for practical applications of such system. More detailed studies in this respect are under progress. References Figure 1 The equilibrium configurations after fully relaxation. White and dark spheres indicate H and Mg atoms, respectively. (a) A top view and (b) a side view of MgH2(001) surface. (c) A top view and (d) a side view of MgH2(110) surface. The twofoldcoordinated H and fourfold-coordinated Mg sites on MgH2(001) surface are labeled 2f and 4f, respectively. The Bridging hydrogen, threefold-coordinated hydrogen, fivefold- and sixfold-coordinated Mg sites are labeled BH, 3f, 5f and 6f, respectively. The white atoms with a cross indicate the two H atoms to be desorpted and recombined into hydrogen molecule. [1]Liang, G.; Huot, J.; Van Neste, A.; Schulz, R. J Alloys Compounds 1999, 292, 247. Zaluska, A.; Zaluski, L.; Strom-Olsen, J.O. Appl.Phys.A: Mater.Sci. Process. 2001, 72, 157. [2] Bazzanella, N.; Checchetto, R. And Miotello, A. Appl.Phys.Letts. 2004, 85, 5212. [3] Au, M. Mater.Sci.&Eng. B 2005, 117, 37. [4] Hanada, N.; Ichikawa, T.; Fujii, H. J.Phys.Chem B 2005, 109, 7188. Label Index MgH2(001) MgH2(110) 1 0.1204 0.115 2 -0.2168 0.146 [5] Greeley, J.; Mavrikakis, M. Nature materials 2004, 3, 810. Vang, R.T.; Honkala, K.; Dahl, S.; Vestergaard, E.K.; Schnadt, J.; Egsgaard.E.; Clausen, B.S.; Norskov, J.K., Besenbacher, F. Nature materials 2005, 4, 160. [6] Yu, R. and Lam, P.K. Phys.Rev.B 1988, 37, 8730. [7] Tsuda, M.; Dino, W.A.; Nakanishi, H. and Kasai, H. J.Phys.Soc.Jpan 2004, 73 2628. [8] Kresse, G.; Furthmuller, J. Comput. Mater. Sci 1996, 6, 15. Kresse, G.; Furthmuller, J. Phys.Rev.B 1996, 54, 11169. 3 -0.0771 0.254 4 0.1566 -0.116 [9] Blochl, P.E. Phys.Rev.B 1994, 50, 17953. Kresse, G.; Joubert, D. Phys.Rev.B 1999, 59, 1758. [10]. Henkelman, J.; Jónsson, H. J. Chem. Phys., 2000, 113, 9978. [11] Henkelman, J.; Uberuaga, B.P. ; Jónsson, H. J. Chem. Phys., 2000, 113, 9901. 5 0.0530 0.087 6 -0.0771 -0.037 Table I Ion Displacements along Z axis after relaxation for MgH2(001) and MgH2(110) surfaces, respectively. Labels refer to Fig.1.(b) and Fig.1.(d). The displacements are in Å, and are from the bulk terminated positions. Acknowledgements We acknowledge generous grants of high-performance computer time from both the Computational Molecular Science cluster computing facility at The University of Queensland and the Australian Partnership for Advanced Computing (APAC) National Facility. The authors also greatly appreciate the financial support by Australian Research Council through the ARC Center for Functional Nanomaterials.