VGs_review
advertisement

Critical Point Drying
1.
2.
3.
4.
Biological specimens
MEMS
Aerogels
Spices
Single film annealed: 16 stages
p
xx / L0 = ( A + B T ) fit
3
2
L0= e / h
xx / L0
A = -0.01 (1)
B = 0.622 (9)
p = 0.390 (3)
2
R0 (K)
R0
1
increasing
4
15.59
16.56
21.54
22.67 (Rc)
17.01
18.30
18.57
19.08
19.68
20.63
PowerLaw fit
23.77
25.01
26.11
28.90
30.83
10
Temperature (K)
60
Single film annealed: 16 stages
W = T{d ln(T)/dT }
0.6
R0(k)
0.5
15.59
19.08
23.77
16.56
17.01
18.30
18.57
19.68
20.63
21.54
22.67
25.01
26.11
28.90
30.83
d ln( (T )
w(T )
d ln(T )
d ln( A BT p )
d ln T
pBT p
A BT p
0.4
0.3
R increasing
0
5
10
15
20
25
30
35
40
45
Temperature (K)
50
55
Scaling collapse for indicated values of R0
(metallic side, recall Rc=22.67 k)
Measurement of Irreversible magnetization
• Field cooled (MFC) and Zero field cooled (MZFC) magnetization.
• ∆M(H,T) = MFC(H,T) – MZFC(H,T) , is the irreversible
magnetization
Field dependence of ∆M(H,T) isotherms
Ni/AlOx multilayers
(Hm(T), ∆Mmax (T))
Increasing T
Hm(T)
→ Magnetic field where maxima of ∆M occures
∆Mmax(T) → Maximum value of ∆M
Scaling collapse of ∆M
(T)
Ni/AlOx multilayers
T
∆M (H,T) = ∆Mmax(T)F(H/Hm)
(T)
Scaling collapse in other materials
3
Ni SD nanoparticles (3nm)
LPCMO
M/ M max
2
FePt nanoparticles (6 nm)
1
Ni MD nanoparticles (12 nm)
Gd thin film (50 nm thick)
0
Cu:Mn (1.5 at %) Spinglass
0
2
4
H/H
m
6
Zotev , Orbach et. all, PRB, 2
Hydrogen Molecules
(Quadrapolar Glass)
The combined 1st and 2nd Laws
The 2nd law need not be restricted to reversible processes:
dU đQ đW
đQr đWr
TdS PdV
• đQ is identifiable with TdS, as is đW with PdV, but only
for reversible processes.
• However, the last equation is valid quite generally, even
for irreversible processes, albeit that the correspondence
between đQ & TdS, and đW & PdV, is lost in this case.
Conversion of Heat to Work (a heat engine)
Efficiency (h*):
Heat reservoir at
temperature T2 > T1
W
W
output
h
Q2 Q2
input
Q2 Q1
Q1
h
1
Q2
Q2
h 1
Q1
Q2
Q2
Heat
Engine
Q heat
W work
both in Joules
W Q2 Q1
Q1
Heat reservoir at
temperature T1 < T2
Q2 Q1
*Don’t
confuse with JouleThomson coefficient
The combined 1st and 2nd Laws
dU TdS PdV dn
U
U
U
dS
dV
dn
S V ,n
V S ,n
n S ,V
U
T
;
S V ,n
U
P
;
V S ,n
U
n
S ,V
If more than one type of particle (constituent) is
added, then
m
dU TdS PdV j dn j
j 1
U
j
n j
k 1 m j
S ,V ,nk
Work and Internal Energy
•Differential work đW is inexact (work not a state variable)
•Configuration work is the work done in a reversible process
given by the product of some intensive variable (y) and the
change in some extensive variable (X).
•đW is the work done on ‘the system’, e.g. đW is positive
when a gas contracts.
•Dissipative work is done in an irreversible process and is
always done ‘on the system’, i.e. đWirr > 0 always.
•Total work (configuration and dissipative) done in adiabatic
process between two states is independent of path. This
leads to the definition of internal energy (state variable).
b
a
dU Ub U a
đW
b
a
ad
Wad
and
dU đWad
Equation of State of an Ideal Gas
•In chapter 1, we used the zeroth law to show that a
relationship always exists between P, V and T.
General form: f (P,V,T) = 0
Example:
PV = nRT
(ideal gas law)
n quantifies the amount of the substance. The units
of R and n are linked such that their product nR
has the dimensions of joules/kelvin.
•If n measured in kilomoles, then R = 8.314 103 J/kilomoleK
•If n measured in moles, then R = 8.314 J/moleK
•Ideal gas law may also be written in intensive form
Pv = RT
v is the specific volume in either m3/mole or m3/kilomole
Equation of State of a Real Gas
Van der Walls’ equation in intensive form:
a
P 2 v b RT
v
100
90
Pressure (arb. units)
80
o
70
60
50
o
12
o
10
o
9
o
8
o
6
o
4
CP
40
11
o
9.5
o
8.5
o
7
o
5
o
3
30
20
10
0
0.1
0.2
0.3
0.4
0.5
Specific volume (arb. units)
0.6
London – van der Waals’ interaction
Etotal = K + V = constant
•So, the addition of particles to a system results in the
addition of kinetic energy to the system which almost
immediately dissipates to the entire system.
•Therefore, the addition of particles changes the
internal energy of the system. The change in the
internal energy dU is proportional to the number of
particles dn that are added. The proportionality
constant is called the ‘chemical potential’.
Thus, we need to modify the combined 1st and 2nd laws:
dU = TdS – PdV + dn
Thermal (kinetic)
Mechanical, chemical (potential)
Properties of Heat
It is the temperature of a body alone that determines
whether heat will flow to or from a body,
“Heat energy is transferred across the boundary of a
system as a result of a temperature difference only.”
•However, this does not necessarily imply that the transfer
of heat to a body will increase its temperature. It may
also undergo a change of state (phase) from e.g. a liquid
to a gas, without a change in temperature.
•Also, if the temperature of a system increases, it does
not necessarily imply that heat was supplied. Work may
have been done on the system. Therefore,
“Heat is the change in internal energy of a system
when no work is done on or by the system.”
Heat Capacity and specific heat
The heat capacity C of a system is defined as:
Q đQ
C lim
T 0 T
dT
•Heat capacity is an extensive quantity.
The specific heat capacity c of a system is:
1 đQ đq
c
n dT dT
•Specific heat is obviously an intensive quantity.
•SI units are J.kilomole-1.K-1.
Heat Capacity and specific heat
Because the differential đq is inexact, we have to specify
under what conditions heat is added.
•
the specific heat cv; heat supplied at constant volume
•
the specific heat cP; heat supplied at constant pressure
đq
cv
dT
v
and
đq
cP
dT
P
Using the first law, it is easily shown that:
đq u
cv
dT
T
v
v
•For an idea gas, the internal energy depends only on the
temperature of the gas T. Therefore,
du
cv
dT
and
T
u u0 cv dT
T0
The Gay-Lussac-Joule Experiment
u
T u
T
cv
v T
v u T v
v u
• Suggests an experiment: measure temperature change of
a gas as a function of volume whilst keeping u fixed; this
will enable us to determine how u depends on v.
T
T1 T0
dv
v0
v u
v1
The Joule coefficient
T
h 0.001 K kilomole m3
v u
• For an ideal gas:
u
h 0, so 0
v T
u u T
Expansivity and Compressibility
Two important measurable quantities:
Expansivity:
1 v
v T P
Compressibility:
1 v
v P T
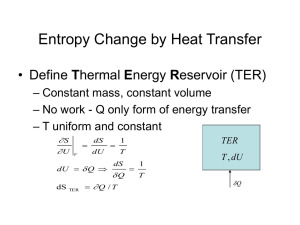
The Second Law of Thermodynamics
• Clausius’ statement: It is impossible to construct a device
that operates in a cycle and whose sole effect is to transfer
heat from a cooler body to a hotter body.
• Kelvin-Planck statement: It is impossible to construct a
device that operates in a cycle and produces no other effect
than the performance of work and the exchange of heat
from a single reservoir.
• Carnot’s theorem: No engine operating between two
reservoirs can be more efficient than a Carnot engine
operating between the same two reservoirs.
The Clausius Inequality and the 2nd Law
Consider the following cyclic process:
P
2
Irreversible
Reversible
1
V
The Clausius Inequality and the 2nd Law
The Clausius inequality leads to the following relation
between entropy and heat:
đQ
dS
T
This mathematical statement holds true for any process.
The equality applies only to reversible processes.
For an isolated system, đQ = 0, therefore
dS 0
or
S S2 S1 0
This leads to the following statement:
• The entropy of an isolated system increases in any
irreversible process and is unaltered in any reversible
process. This is the principle of increasing entropy.
The Carnot Cycle
1.
2.
3.
4.
ab isothermal expansion
bc adiabatic expansion
cd isothermal compression
da adiabatic compression
1.
2.
3.
4.
W2 > 0, Q2 > 0 (in)
W' > 0, Q = 0
W1 < 0, Q1 < 0 (out)
W'' < 0, Q = 0
T1
h 1
T2
• Stirling’s engine is a good approximation to Carnot’s cycle.
Cooling via Work (a Refrigerator)
Environment at
temperature T2 > T1
Q2
W
Refrigerator
Q1
Refrigerator Inside,
temperature T1 < T2
Coefficient of
Performance (c):
Q1 Q1
c
W W
Q1
Q2 Q1
T1
T2 T1
Available energy
T1 đW
h 1
T2 đQ
T2 > T1
đQ
đW
M
Available energy in a reversible
cycle:
(1 – T1/T2)đQ
Unavailable energy:
T1đQ/T2
T1 < T2
Efficiency = h
There exists no process that can
increase the available energy in
the universe.
Entropy changes in reversible processes
đqr du Pdv
đqr du P
ds
dv
T
T T
Various cases:
•
For an ideal gas quite generally:
T2
v2
s2 s1 cv ln R ln
T1
v1
Entropy changes in reversible processes
đqr du Pdv
đqr du P
ds
dv
T
T T
Various cases:
•
Adiabatic process: đqr = 0, ds = 0, s = constant. A reversible
adiabatic process is isentropic. THIS IS NOT TRUE
FOR AN IRREVERSIBLE PROCESS!
•
Isothermal process:
s2 s1 ds
2
1
đqr qr
T
T
Entropy changes in reversible processes
đqr du Pdv
đqr du P
ds
dv
T
T T
Various cases:
•
Isochoric process: We assume u = u(v,T) in general, so
that u = u(T) in an isochoric process. Therefore, as in
the case for an ideal gas, du = cvdT. Thus,
s2 s1
2
1
T2
dT
cv
cv ln ,
T
T1
provided cv is independent of T over the integration.
Entropy changes in reversible processes
đqr du Pdv
đqr du P
ds
dv
T
T T
Various cases:
•
Isothermal (and isobaric) change of phase:
l
s2 s1 ,
T
where l is the latent heat of transformation.
Entropy changes in reversible processes
đqr du Pdv
đqr du P
ds
dv
T
T T
Various cases:
•
Isothermal (and isobaric) change of phase:
l
s2 s1 ,
T
where l is the latent heat of transformation.
Entropy changes in reversible processes
đqr du Pdv
đqr du P
ds
dv
T
T T
Various cases:
•
Isobaric process: More convenient to deal with enthalpy
dh du Pdv vdP
đqr dh v
ds
dP
T
T T
h = h(P,T) in general, so that h = h(T) in isobaric process.
s2 s1
2
1
T2
dT
cP
cP ln ,
T
T1
provided cP is independent of T over the integration.
The Tds equations
The Joule-Thomson Experiment
0
w1 Pdv
Pv
1
1 1
v1
v2
w2 P2 dv P2 v2
0
w w1 w2 P2 v2 Pv
1 1
u u1 u2
u1 Pv
1 1 u2 P2 v2
or
h1 h2
• Thus, a throttling process occurs at constant enthalpy.
• The experiment then involves throttling the gas for
different values of P2. If h depends on P, then T will
change during a constant h throttling process (next slide).
• This contrasts the previous experiment where the search
was for a change in T during a constant u expansion.
Enthalpy
When considering phase transitions, it is useful to define a
quantity h called ‘enthalpy’
h u Pv
•Because h depends only on state variables, it too must be
a state variable – hence its usefulness.
•When a material changes phase (e.g. from a solid to a
liquid) at constant temperature and pressure, latent heat l
must be added. This heat of transformation is related
simply to the enthalpy difference between the liquid and
solid
l u2 Pv2 u1 Pv1 h2 h1
Enthalpy and specific heat
•The specific heat is not defined at any phase transition
which is accompanied by a latent heat, because heat is
transferred with no change in the temperature of the
system, i.e. c = ∞.
•However, enthalpy turns out to be a useful quantity for
calculating the specific heat at constant pressure
đq
h
cP
dT
T
P
P
•For an ideal gas, the enthalpy depends only on the
temperature of the gas T. Therefore,
dh
cP
dT
and
T
h h0 cP dT
T0
The Gibbs function And Chemical Potential
Using Euler’s theorem, one can now define an absolute
value for the internal energy of a system
m
U ST PV j n j
Thermal
(kinetic)
j 1
Mechanical
(potential)
Chemical
(potential)
From this definition, one can easily show that
m
G jnj
j 1
m
and
dG T ,P j dn j
j 1
From the above, one readily sees that = G/n for a
system with only one constituent, i.e. = g. This is not
true for mixtures or multi-constituent systems.
Enthalpy
When considering phase transitions, it is useful to define a
quantity h called ‘enthalpy’
h u Pv
•Because h depends only on state variables, it too must be
a state variable – hence its usefulness.
•When a material changes phase (e.g. from a solid to a
liquid) at constant temperature and pressure, latent heat l
must be added. This heat of transformation is related
simply to the enthalpy difference between the liquid and
solid
l u2 Pv2 u1 Pv1 h2 h1
Enthalpy and Pressure
h
T h
T
cP
P T
P h T P
P h
• Thus, we see the connection between the physics.
• The main difference is that enthalpy is relevant during
constant pressure processes, whereas internal energy is
relevant during constant volume processes.
T
The Joule-Thomson coefficient:
0 for most gases
P h
• For an ideal gas:
h
0, so 0
P T
h h T
The Tds equations
T
P
Tds cv dT T
dv
dv cv dT
T v
v
Tds cP dT T
dP cP dT Tv dP
T P
s s v, T
s s P, T
cv
cP
T
T
Tds cP
dv
dP s s v, P
dv cv
dP
v
v P
P v
These equations give:
•heat transferred (đq = TdS) in a reversible process;
•the entropy by dividing by T and integrating;
•heat flow in terms of measurable quantities;
•difference in specific heat capacities, cP, , , T, etc..;
•relationships between coordinates for isentropic processes.