Leukemias
advertisement

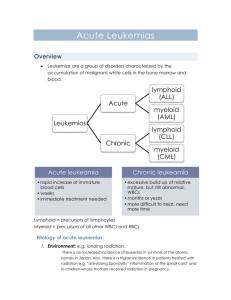
Jan Żeromski Pathology 2010/2011 Malignant proliferation of white cells of the hematopoietic system with infestation of blood and usually bone marrow. Lymphoid and myeloid leukemias Hematopoietic stem cells differentiating to progenitor ones, either myeloid or lymphoid lineage Chromosomal abnormalities, most commonly translocations, Inherited genetic defects (Down syndrome, Bloom syndrome, ataxia telangiectasia Some viruses: Epstein-Barr (EBV), herpesvirus -8, HTVL1 etc. Environmental factors: chronic infection, Helicobacter pylori, radiation therapy, chemotherapy Precursor leukemias - acute lymphoblastic l. - pro B - pre B (common) Peripheral ones - chronic lymphocytic l. - prolymphocytic l. - hairy cell l. - plasma cell myeloma B-cell origin Precursor leukemias - acute lymphoblastic l. Peripheral ones (T and NK) - chronic lymphocytic l. - large granular lymphocytic l. - Sezary syndrome - NK-cell leukemia T-cell origin Definition: the set of differentiation antigens expressed on a given cell type Lymphoid and myeloid cell subsets in various stages of maturation have distinct immunophenotype Differentiation antigens are classified according to cluster determinants (CD) system CD classification covers more than 300 antigenic epitopes searched by monoclonal antibodies Each leukemia type has characteristic set of CD markers, what constitutes nowadays current classification hematopoietic malignancies and the basis of diagnosis B cell lineage antigens differentiation Clonality – crucial difference between neoplastic and normal lymphocytes Unique feature of lymphocytes – antigen receptor Antigen receptor gene rearrangement determines antigen specificity of lymphocytes Rearrangement precedes neoplastic transformation Thus daughter cells from malignant cell progenitor express the same antigen receptor (either T or B) – form identical cell clone Acute Lymphoblastic Leukemia • Most cases are Tdt positive • Most express CD10 (common ALL antigen) • Most are “pre-B cell” phenotype • 15-20% T-cell lineage • 5% B-cell phenotype Acute myelogenous leukemias (8 types) Myelodysplastic syndromes (MDS) Chronic myeloprolifeative disorders: - chronic myelogenous leukemia polycythemia vera essential thrombocytosis primary meylofibrosis M0 AML – non-differentiated, incidence 2-3% M1 AML – almost without differentiation, 20% M2 AML – myeloid maturation of granuloc.30-40% M3 AML – hypergranular promyelocytes, 5-10% M4 AML – cells of both myelocytic (min.20%) and monocytic origin (<30%), 15-20% M5 AML – Monoblasts (M5a) and monocytes (M5b), 10% M6 AML – dysplastic erythroid precursors and myeloblasts, 5% M7 megakaryocytic leukemia – blasts of megakaryoc.lineage predominate, 1% Number of blasts in all: < 20% Myeloid cell lineage antigens differentiation Chromosomal translocation is fairly common and may be distinct for given subtype such as t(15;17) in M3 AML Some leukemias do no fit to current classifications and are described on the basis of several cytogenetic defects such as gene fusion, deletion, insertion etc. In lymphoid tumors antigen receptor gene rearrangement is earlier than neoplastic transformation. Thus, tumorous gene products (immunoglobulins or Tcell receptors) are identical in contrast to normal lymphocytes expressing plethora of various antigen receptors. It turned out to be of diagnostic value. General: symptoms are more evident in acute than in chronic forms , but do not differ much in lymphoid vs. myeloid leukemias, In chronic types the onset is usually insidious: mild anemia, weakness, easy fatigue, weight loss, anorexia Enlargement of spleen is common in both types often associated with left upper quadrant pain, Acute leukemias manifest by abrupt stormy onset: fever, infection, bleeding, bone pain, hepatosplenomegaly, cns manifestations such a vomiting, headache, nerve palsies. Chronic Leukemia • Often discovered because of an abnormal lab or an abnormal physical examination • Severe cytopenias characteristic of acute leukemia are seldom present at time of diagnosis Fairly good in precursor B-cell leukemias in childhood In chronic B-cell leukemias life expectancy may be comparable to normal one, provided that there is no cns involvement Acute myeloid leukemias have usually unfavourable prognosis (15-30% remain free of disease for 5 years), In chronic myelogenous leukemia supplementation of blood and proper chemotherapy allow fro 4-5 years survival, but the end is almost always fatal. Clinical – pancytopenia, normal lymph nodes and splenomegaly Incidence –mostly middle age males (2% of lymphomas) Origin – mature B cells but with peculiar projections best seen in phase-contrast microscope. They are pan B, CD5-, CD25+, CD11c+, CD103+ Infiltration of spleen, bone marrow and liver Accompanying infections common often with M0 AML – non-differentiated, incidence 2-3% M1 AML – almost without differentiation, 20% M2 AML – myeloid maturation of granuloc.30-40% M3 AML – hypergranular promyelocytes, 5-10% M4 AML – cells of both myelocytic (min.20%) and monocytic origin (<30%), 15-20% M5 AML – Monoblasts (M5a) and monocytes (M5b), 10% M6 AML – dysplastic erythroid precursors and myeloblasts, 5% M7 megakaryocytic leukemia – blasts of megakaryoc.lineage predominate, 1% Number of blasts in all: < 20% Incidence: middle age, older adults and children Bone marrow: normal cells are replaced but undifferentiated blasts with some myeloid features. They have blocked maturation, increased survival and the replication rate lower than normal myeloid progenitors, Genetic: several chromosomal rearrangements disrupting genes encoding transcription factors needed for normal myeloid maturation, Lab findings: anemia, neutropenia, thrombocytopenia Flow cytometry: due to poor cell granularity blasts have very low site scatter. They usually show myeloid-specific surface markers (CD13, CD33) and markers of progenitor cells (CD34, CD117), but various departures from the above are common, Clinical: fatigue, spontaneous bleedings, infections, often caused by opportunistic pathogens such as fungi or Pseudomonas. Organomegaly of liver, spleen, infiltration of skin, gingiva by normal monocytes is often seen. Petechiae, Purpura Hematoma, Joint bl. Disease of adults (25 – 60 yrs), more common in women, Bone marrow – highly cellular, mainly maturing granulocyte precursors, increased numbers of megakaryocytes Molecular – translocation of BCR gene (chromosome 9) to ABL gene (on chromosome 22) The resultant BCRABL fusion gene encodes protein with tyrosine kinase activity. So-called Philadelphia chromosome seen in karyotyping studies is the reflection of (9;22) translocation Clinical – slow progression up to 5 years, afterwards some 50% of patients enter „accelerated phase” with appearance of blasts, resembling acute leukemia. BCR-ABL translocation Chronic Myelogenous Leukemia Cell origin: multipotent myeloid stem cell Bone marrow: increased production of erythroid, granulocytic and megakaryocytic maturing cell types Blood: erythrocytosis, granulocytosis, thrombocytosis, increased hematocrit and red cell mass Clinical: insidious onset in late middle age (above 60 yrs). Patients suffer from headache, dizziness, hypertension, gastrointestinal abnormalities, gout due to high cell turnover. Thrombosis episodes are common. Final fate: either „spent phase” leading to marrow myelofibrosis with extramedullary hematopoesis or terminal AML Definition: clonal cell disorders marked by maturation defects, ineffective hematopoiesis and increased risk of AML (10-40%) Two distinct types: idiopatic (primary) MDS in older people with insidious course. Often discovered incidentally on routine testing. therapy-related MDS, due to previous chemo-or radiation therapy, usually 2 to 8 years after, Bone marrow: several distorted cells of myeloid lineage showing various abnormalities. Myeloblasts may be increased, but below 20%. If higher – it is already leukemia, Survival: primary MDS-9-29 mo, therapy-related – 4 to 8 mo.