Immunomodulation and cancer:
Different relationships across
diseases and disease states?
Rafael Ponce
Sept 27, 2012
Immunomodulation and cancer
Virus
Immune
function
• Inflammation, immune activation
• Used by host to eliminate malignant cells
(immunosurveillance)
• Used by tumor to create a permissive
environment for growth/development
• Drives lymphoma development (chronic B
cell activation)
• Immunosuppression
• Used by tumor to escape surveillance
• Increased risk of oncogenic virus activity
• Increased risk of unresolved infection
Tumor
• Immune escape mechanisms
• Perception of ‘self’ in the absence of
‘danger’, Ignorance: Peripheral tolerance,
Down-regulation of MHC class I
• Active immunosuppression, induced
tolerance
Need to break tolerance
• Evolve under selective pressure of immune
response to acquire mechanisms for immune
escape
Immune status in the tumor microenvironment drives balance of response
(tolerance vs immunity)
Immunity and cancer paradigms
1.
2.
3.
4.
Immunosurveillance model
Inflammation model
Lymphomagenesis model
Oncogenic virus model
All models have experimental and epidemiological support
How can we understand the role of immunity and cancer for specific cases?
1. Immunosurveillance model
• Innate and adaptive immune cells protect the host
from transformed cells (elimination)
– NK, NKT, CD4+ T cells, CD8+ T cells, DC
• Transformed cells can adapt to immune surveillance,
establish a fight for dominance (equilibrium)
• Transformed cells overcome immune surveillance,
develop into clinically apparent tumors (escape)
1. Immunosurveillance model
1. Immunosurveillance model
Cancer immunosurveillance
Tumor supportive
environment
M
IL-12
M
PGE2
IL-23
VEGF-C/D
TH17
IL-6
IL-1b
TGF-b TNF-a
Anti-tumor adaptive
immune response
B cell
IDO
TGF-b
IL-10
PGE2
PD-L1
B7-H1
B7-H3
B7x
HLA-G
HLA-E
Tumor
Parenchyma
Perforin
TRAIL
NK Cell
IL-12, IFN-g, a-GalCer
NKT
Cell
IFN-g
Perforin
DC
Treg
IL-35 IDO
IL-10 TGF-b
PD-L1 PGE2
IL-13, IL-6
TGF-b
MDSC
Imm DC
Tumor
escape
pDC
Tumor elimination
2. Inflammation model
• Chronic inflammation can
– induce cell transformation (reactive
oxygen/nitrogen spp),
– promote cell proliferation and increase the risk of
spontaneous mutations, and
– create a permissive environment for tumor
growth and spread
2. Inflammation model
Also, Mantovani et al (2008) Nature 454:436-444
3. Lymphomagenesis model
• B cell lymphomas occur at different steps of B-cell development and
represent their malignant counterpart
• Lymphomas arise from errors occurring at hyper-mutable stages of B cell
development
– Genetic hallmark is chromosomal translocations resulting from aberrant
rearrangements of IG and B(or T) cell receptor genes
– Leads to inappropriate expression of genes at reciprocal breakpoints that
regulate a variety of cellular functions
• gene transcription, cell cycle, apoptosis, and tumor progression
• Lymphomas promoted by chronic B cell activation (infection, alloantigen (graft),
self-antigen (autoimmunity))
3. Lymphomagenesis model
B- cell development
3. Lymphomagenesis model
B- cell development requires DNA recombination
B- cell development requires DNA
recombination
V(D)J recombination
Process for assembling
gene segments coding
variable region of
antibody molecule to
generate Ab diversity
Class switch
recombination
Process for altering
effector activity of
heavy chain via
recombination of Fc
heavy chain
Somatic
hypermutation
Process for altering
antibody specificity via
point mutations,
deletions, duplications
Errors arising in hyper-mutable stages of B-cell
development drives lymphoma
Klein and Dalla-Favera (2008) Nat Rev Immunol 8:22
3. Lymphomagenesis model
4. Oncogenic virus model
• Innate and adaptive immunity protects the host from
active infection by oncogenic viruses
– NK cells, CD8+ T cells, CD4+ T cells, granulocytes, DC
• Seven identified human oncogenic viruses
–
–
–
–
–
–
EBV: B cell lymphoma
Hepatitis B, C viruses: hepatocellular carcinoma
HTLV-1: T cell leukemia/lymphoma
HHV8 (KSHV): Kaposi’s sarcoma
HPV: Cervical cancer, anogenital cancers, oropharyngeal
cancers
Merkel cell polyomavirus: Merkel cell carcinoma
Role of oncogenic viruses
• Variable attribution of cancer to oncoviruses
–
–
–
–
–
–
HPV and cervical cancer (~100%)
CNS lymphoma and EBV (HIV patients, 100%)
Merkel cell polyoma virus and MC carcinoma (80%)
HTLV-1 and Adult T cell leukemia/lymphoma (?)
HHV8 and Kaposi’s sarcoma (~100%)
EBV and Lymphoma (2 to >90%)
4. Oncogenic virus model: EBV
B-cell transformation by EBV
Relating paradigm to cancer in patient
populations with altered immunity
• Which patient populations provide useful
information?
– Congenital (Primary) immunodeficiency
– Organ transplant recipients
– Acquired immunodeficiency (HIV)
– Autoimmunity
• What forms of cancer prevail in these
populations?
Grulich et al (2007) Lancet 370:59
Relative risk of cancer with
immunomodulation
RR
1
>1-3x
5-10x
10-20x
>20x
HIV/AIDS
(CD4+)
Breast, Prostate
Colon/rectum
Ovary
Thyroid
Leukemia, Lip,
Stomach, Nonmelanoma skin,
Oro-pharynx
Gynecological
cancers
Liver
Vulva/vagina
Hodgkin’s
NHL
Anal cancer
Kaposi’s sarcoma
Organ
transplant
Breast, Prostate
Ovary, Brain,
Testes
Stomach
Cervix
Oro-pharynx
Hodgkin’s
Thyroid
NHL
Kidney
Penis
Kaposi’s sarcoma
Non-melanoma
skin
Lip
Genital cancers
Breast (CVID)
Breast (AT)
Stomach (CVID)
NHL (CVID, SCID,
AT, WAS, XLD)
Stomach (XLA)
Leukemia (AT,
WAS)
NHL (RA)
Other solid organ
(RA)
Leukemia (RA)
Hodgkin’s (RA)
NHL (Sjogren’s,
SLE, Celiac)
T cell lymphoma
(AHA, celiac
disease)
1° Immunodeficiency
Autoimmunity
AHA: Autoimmune hemolytic anemia; CVID: Common variable immunodeficiency; XLA: X-linked agammaglobulinemia
SCID: Severe combined immunodeficiency; AT: Ataxia telangiectasia; WAS: Wiscott-Aldrich syndrome; XLD: X-linked lymphoproliferative disorder
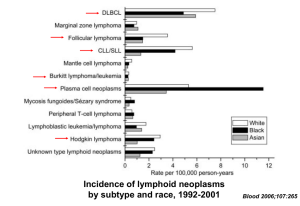
EBV differentially contributes to lymphoma burden across patient populations
Disease
Lymphoma with no known immunosuppression
% EBV+ Tumors
2-10%
>90%
(Kamel et al., 1999; Hoshida et al.,
2007)
(Macsween et al., 2003; Swerdlow,
2003; Young et al., 2003; ThorleyLawson et al., 2004; Young et al., 2004;
Balandraud et al., 2005)
(Macsween et al., 2003; Young et al.,
2003; Young et al., 2004)
(Macsween et al., 2003)
50%
(Young et al., 2004)
Hodgkin’s lymphoma
40-50%
80%
Burkitt’s lymphoma (developed world)
15-25%
NHL
HIV patients
Primary
Immunodeficiency
RA Patients
Post-transplantation
(<1yr)
Post-transplantation
(>1yr)
NHL
Burkitt’s
CNS Lymphoma
Lymphoma/BPLD¶
Lymphoma
Lymphoma (mucosalassociated)
Citation
28-66%
25%
100%
31%#
0%
0%
2%
3%
15%
27%
11%
26%
17%
12%
(Rabkin, 2001; Macsween et al., 2003;
Balandraud et al., 2005)
(Macsween et al., 2003)
(Rabkin, 2001; Macsween et al., 2003)
(Filipovich et al., 1994)
(Gompels et al., 2003)
(Cunningham-Rundles et al., 2002)
(Kamel et al., 1999)
(Staal et al., 1989)
(Mariette et al., 2002)
(Hoshida et al., 2007)
(Askling et al., 2005)
(Dawson et al., 2001)
(Baecklund et al., 2003)
(Baecklund et al., 2006)
Relating paradigm to cancer in patient
populations with altered immunity: A proposal
1. Is cancer associated with oncogenic virus etiology
identified at increased rates?
– What proportion of tumors evidence viral DNA?
2. Is there evidence/risk of inflammation?
–
–
Unresolved infection?
Autoimmunity?
3. Are pathways associated with tumor antigen
detection and adaptive immunity affected?
Which paradigm explains cancer in patient populations
with altered immunity?
RR
HIV/AIDS
(CD4+)
Organ
transplant
1° Immunodeficiency
Autoimmunity
5-10x
10-20x
Gynecological
Hodgkin’s
cancers
4, 1
Liver
4/1?
Hodgkin’s
Thyroid
Breast (AT)
4, 3
1
NHL
Kidney
Penis
>20x
3, 4
NHL
3, 4
Anal cancer
4, 1
Kaposi’s sarcoma 4
4, 3 Kaposi’s sarcoma 4
1 Nonmelnma skin 1
4 Lip
1, 4
Genital cancers 4
--, 1 Stomach (CVID) 2 NHL
Stomach
Leuk (WAS, AT)
NHL
3 (4?)
T cell lymphoma ?
3
2
---
1. Immunosurveillance model
2. Inflammation model
3. Lymphomagenesis model
4. Oncogenic virus model
So what does this tell us?
• Risk of immunomodulation and cancer differ across
patient populations
– Nature of immunomodulation
• Which pathways?
• How many are affected? [Remove redundancy (immunologic
reserve)]
– Underlying patient status
• Nature of inciting antigen
• Concomitant unresolved infection, autoimmunity
• Contributing conditions (AT/DNA repair error)
• Challenges broad generalizations
Case example: Treatment of RA
• Use of anti-TNFs associated with increased lymphoma risk
(labels)
• Available epidemiology data suggests more severe RA
associated with greater background lymphoma risk (not
treatment related)
– Question: Is lymphoma increasing in RA patients treated with antiTNFs? Is this related to disease severity or infection?
Test lymphomas from RA patients with and without clinical history of
anti-TNF use for presence of EBV
Similar EBV rates (as
RA patients)
Use of anti-TNFs is not increasing EBV-mediated tumors
(increase anti-TNF use to suppress autoimmune-mediated
lymphoma)
High rate of EBV (greater than
that for RA patients)
Use of anti-TNFs increasing rate of virally-related
tumors (maintain warning label)
Conclusions
• Our ability to address concerns regarding immunomodulation
and cancer depends on our ability to articulate discrete,
experimentally evaluable hypotheses
• As we move from broad-spectrum immunomodulation to
targeted immunotherapies, we will need to define
experimental tools that address specific needs
• A combination of mechanistic studies, clinical data, and
epidemiology results will be necessary to ‘validate’ and refine
our models