Continuous-Flow Study and Scale-up of Conventionally
Difficult Chemical Processes
by
Nikolay Zaborenko
B.S. Chemical Engineering, Rutgers University, 2004
M.S. Chemical Engineering Practice, Massachusetts Institute of Technology, 2007
Submitted to the Department of Chemical Engineering
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Chemical Engineering
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
May 2010
© 2010 Massachusetts Institute of Technology. All rights reserved
Author
Department of Chemical Engineering
June 1, 2006
Certified by
Klavs F. Jensen
Warren K. Lewis Professor of Chemical Engineering
Professor of Materials Science and Engineering
Thesis Supervisor
Accepted by
William M. Deen
Carbon P. Dubbs Professor of Chemical Engineering
Chairman, Committee for Graduate Students
Continuous-Flow Study and Scale-up of Conventionally
Difficult Chemical Processes
by
Nikolay Zaborenko
Submitted to the Department of Chemical Engineering on May 20, 2010
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Chemical Engineering
Abstract
Microfluidic systems provide valuable tools for exploring, studying, and optimizing
organic syntheses. The small scales and fast transport rates allow for faster experiments and
lower amounts of chemicals to be used, reducing costs and increasing safety. Additionally,
continuous flow processes allow for a large number of experiments to be performed after a
single setup. These advantages were exploited to enable continuous-flow study of chemical
syntheses that are hazardous or difficult to perform by conventional methods and of applying
the acquired knowledge toward improvement of industrial processes at large scales. Using
silicon semiconductor microfabrication techniques, microdevices have designed and
produced to address various challenges in continuous-flow reaction study and synthesis,
enabling operation at reaction conditions not easily obtained in batch setups or on
macroscopic scale.
Several model reactions and systems were selected for study and/or augmentation.
Silicon micromixers were designed and microfabricated to ensure low-pressure-drop
millisecond-scale mixing of liquid streams. The micromixers were used to perform a
quantitative kinetics and scale-up study of the direct two-step synthesis of sodium
nitrotetrazolate by a Sandmeyer type reaction via a reactive diazonium intermediate. The use
of continuous-flow microsystems significantly reduced the typically high explosion hazard
associated with the energetic product and intermediate.
An epoxide ring opening reaction was augmented and kinetics of the reaction were
rapidly obtained using a silicon microreactor at high temperatures and pressures,
demonstrating microreactor utility for rapid reaction space profiling, as well as the use of
continuous flow to easily study and sample reaction conditions not readily accessible in
batch. Scale-up was demonstrated using obtained kinetics. Synthesis steps of two
pharmaceutical APIs were thus studied and greatly accelerated, which may be useful for
considerations of continuous manufacturing.
Finally, a system has been designed and studied to enable microfluidic study of solidsforming reactions such as an organic coupling reaction with inorganic salt byproduct
precipitate. Conventionally, these solids render such reactions difficult to study in
microreactors, which limits the types of chemistries that could be investigated and improved
using microfluidic technology. To minimize these constraints, the formation of solids in flow
was systematically studied, and a combination of reactor design and application of acoustic
forces to effect solid agglomerate disruption was used to allow slurries with relatively large
amounts of solids to flow through microchannels.
Thesis Supervisor: Klavs F. Jensen
Title:
Warren K. Lewis Professor of Chemical Engineering
Professor Materials Science and Engineering
2
To my parents
Tatyana and Yakov
with love
3
Acknowledgments
First and foremost, I am thankful to Klavs for his help, guidance, and advice, as well
as the encouraging and supporting environment he provided. He has been an excellent
thesis advisor, always finding time in his increasingly busy schedule to discuss my work,
address my concerns, and offer suggestions. I still marvel at the depth and breadth of his
technical knowledge, and the uncanny ability to unerringly select a book from his library
and flip to a specific page in answer to nearly any technical question.
Many thanks go to members of my thesis committee, Professors Bill Green, George
Stephanopoulos, and Rick Danheiser. Their valuable comments and suggestions always
helped steer me in the right direction. I also wish to thank Professors Steve Buchwald
and Tim Jamison for the numerous chemistry discussions.
I am deeply grateful to the staff of Microsystems Technology Laboratories, especially
Dave Terry, Dennis Ward, Bob Bicchieri, Kris Payer, and Kurt Broderick, for making
sure I actually end up with working microreactors. For all of the compression chucks and
for providing oft-needed sanity checks, I wish to thank Peter Morley. Sincere thanks, as
well, to Joan, Alina, Katie, and Christine for all the paperwork and scheduling help.
I am greatly indebted to my many collaborators on numerous projects, particularly
Edward Murphy and Jason Kralj on the NaNT study, Matthew Bedore on the epoxide
aminolysis, and Ryan Hartman and John Naber on the solids handling project. I would
also like to thank the many people I’ve worked with over these years, especially Joseph
Martinelli, Hemant Sahoo, Jonathan McMullen, Kevin Nagy, and Patrick Heider. Many
thanks also to Saif Khan, Michiel Kreutzer, Kishori Deshpande, Vicki Dydek, Chris
Marton, and Andrea Adamo for being frequent sounding boards and sources of advice
I would especially like to thank Eddie, Jason, Saif, Hemant, Kishori and Jamil for
first welcoming me into the group. Not only did you impart upon me your invaluable
wisdom and experience, but you made it fun.
For life outside of the walls of 66, thank you to the poker/Muddy/cookout crew: Jon,
Chris, Vicki, Zac, Jason, Kelly, Wayne, Mike, and Ryan.
I dedicate this thesis to my parents, Tatyana and Yakov. I will always be grateful for
their love and support, motivation and encouragement, and the carloads of good food.
4
Table of Contents
Chapter 1.
Introduction......................................................................... 12
1.1
Background – Microchemical Systems ............................................. 12
1.2
Motivation............................................................................................ 13
1.3
Thesis Objectives and Outline ........................................................... 15
Chapter 2.
2.1
Wet-Etch Fabrication......................................................... 17
Silicon Microreactor Etching............................................................. 18
2.1.1
Plasma Etching ..................................................................................... 19
2.1.2
Potassium Hydroxide Etching .............................................................. 21
2.1.3
Acidic Wet Etching............................................................................... 26
2.2
Reactor Design and Fabrication........................................................ 29
2.2.1
KOH-Etched Reactors .......................................................................... 29
2.2.2
HNA-Etched Reactors .......................................................................... 39
2.3
Results .................................................................................................. 42
2.3.1
KOH-Etched Microreactors.................................................................. 42
2.3.2
KOH-Etched Mesoscale Reactors ........................................................ 45
2.3.3
HNA-Etched Mesoscale Reactors ........................................................ 50
2.4
Chapter 3.
Conclusions.......................................................................................... 53
Interdigitated Micromixers ............................................... 54
3.1
Continuous Micromixing ................................................................... 55
3.2
Micromixer Design ............................................................................. 56
3.2.1
Three-Wafer Stack................................................................................ 56
3.2.2
Two-Wafer Stack.................................................................................. 59
3.3
Micromixer Fabrication and Packaging........................................... 60
3.3.1
First-Generation Devices ...................................................................... 60
3.3.2
Second-Generation Devices.................................................................. 63
3.3.3
Compression Packaging........................................................................ 64
3.4
Micromixer Qualification................................................................... 65
3.4.1
Micromixer Characterization Method .................................................. 65
3.4.2
Characterization Experimental Setup ................................................... 66
5
3.4.3
Characterization Results and Discussion.............................................. 68
3.4.4
Cooling of Three-Stream Micromixer .................................................. 70
3.5
Chapter 4.
Conclusion ........................................................................................... 71
Sodium Nitrotetrazolate Kinetics...................................... 72
4.1
Motivation............................................................................................ 73
4.2
Sodium Nitrotetrazolate Synthesis Description ............................... 74
4.3
Micromixer-Based System Design..................................................... 75
4.3.1
DHT 2 Formation Study ....................................................................... 75
4.3.2
NaNT 3 Formation Study ..................................................................... 77
4.3.3
Scale-up to NaNT 3 production ............................................................ 82
4.4
Reaction Kinetics Evaluation and Scale-up...................................... 83
4.4.1
Kinetic evaluation of DHT 2 formation................................................ 83
4.4.2
Kinetic evaluation of NaNT 3 formation.............................................. 86
4.4.3
Scale-up to NaNT 3 production ............................................................ 92
4.5
Chapter 5.
Conclusion ........................................................................................... 95
Epoxide Aminolysis ............................................................ 96
5.1
Motivation............................................................................................ 97
5.2
Epoxide Aminolysis Synthesis............................................................ 99
5.3
Microchemical System Design ......................................................... 102
5.3.1
Microreactor Design ........................................................................... 102
5.3.2
Continuous-Flow System Setup.......................................................... 104
5.3.3
Heat Transfer Analysis ....................................................................... 105
5.4
Reaction Screening and Profiling.................................................... 109
5.4.1
Experimental Setup and Operation ..................................................... 109
5.4.2
Results and Discussion ....................................................................... 111
5.4.3
Model Chemistries.............................................................................. 112
5.4.4
Application to Pharmaceutical Compounds ....................................... 118
5.5
Kinetic Evaluation of Epoxide Aminolysis ..................................... 122
5.5.1
General Procedure............................................................................... 122
5.5.2
Kinetic Evaluation of Bisalkylation.................................................... 123
5.5.3
Kinetic Evaluation of Primary Aminolysis......................................... 126
6
5.6
Reaction Scale-up.............................................................................. 130
5.7
Conclusion ......................................................................................... 135
Chapter 6.
Solids Handling in Flow ................................................... 137
6.1
Introduction....................................................................................... 138
6.2
Palladium-catalyzed Amination ...................................................... 139
6.3
Aminolysis Experimental Procedure .............................................. 141
6.4
Microreactor Design ......................................................................... 142
6.4.1
Initial Microreactor Evaluation........................................................... 142
6.4.2
Spiral-channel Microreactor Designs ................................................. 146
6.5
Fluoropolymer Surface Modification.............................................. 155
6.6
Application of Acoustics................................................................... 158
6.6.1
Acoustic Irradiation ............................................................................ 158
6.6.2
Integration of Acoustics with Silicon Microreactors .......................... 162
6.6.3
Effect of Acoustics on Reaction Parameters....................................... 163
6.6.4
Effect of Acoustics on Solids Handling.............................................. 167
6.7
Chapter 7.
Conclusions........................................................................................ 170
Summary and Future Outlook ........................................ 173
7.1
Thesis Contributions......................................................................... 173
7.2
Suggestions for Future Work and Outlook .................................... 175
References
.. .......................................................................................... 177
Appendix A Fabrication Details............................................................ 193
7
List of Figures
Figure 2.1. Etch profiles formed by RIE and DRIE . ...................................................... 19
Figure 2.2. Scanning electron microscopy images of DRIE-etched side wall................. 20
Figure 2.3. Illustrations of features etched by KOH........................................................ 22
Figure 2.4. Illustration of a 90º turn aligned to <100> direction ..................................... 23
Figure 2.5. Photographs of KOH-etched 90º and 180º corners ....................................... 23
Figure 2.6. Illustrations of HNA-etch profiles with and without agitation...................... 28
Figure 2.7. Corner compensation features ....................................................................... 31
Figure 2.8. KOH-etched microreactor illustration and photograph................................. 31
Figure 2.9. Illustration of the layout of the Goldilocks devices on a wafer..................... 32
Figure 2.10. KOH-etched meso-reactor illustration and photograph .............................. 34
Figure 2.11. Goldilocks compression chuck.................................................................... 38
Figure 2.12. Meso-reactor compression chuck and fully assembled meso-reactor ......... 39
Figure 2.13. Spiral mesoscale silicon reactor layout and actual device........................... 40
Figure 2.14. Goldilocks reactor set. ................................................................................. 43
Figure 2.15. Etched convex <110>-aligned corners after equal etch times..................... 43
Figure 2.16. SEM image of KOH-etched reactor bottom................................................ 44
Figure 2.17. Reactor flaws in KOH etching .................................................................... 44
Figure 2.18. RTDs of the KOH-etched meso-scale reactor at 0.5 mL/min. .................... 47
Figure 2.19. RTDs of the KOH-etched meso-scale reactor at 1.0 mL/min. .................... 47
Figure 2.20. Thermally packaged meso-scale reactor with temperature profile.............. 48
Figure 2.21. RTDs of the HNA-etched meso-scale reactor at 0.5 mL/min. .................... 52
Figure 2.22. RTDs of the HNA-etched meso-scale reactor at 1.0 mL/min. .................... 53
Figure 3.1. Interdigitated silicon micromixer .................................................................. 57
Figure 3.2. Two-stream and three-stream interdigitated silicon micromixers................. 60
Figure 3.3. First-generation micromixer lithography masks ........................................... 61
Figure 3.4. First-generation micromixer fabrication process........................................... 61
Figure 3.5. Second-generation micromixer fabrication process. ..................................... 63
Figure 3.6. Three-stream mixer compression chuck........................................................ 65
Figure 3.7. Experimental setup for the Villermaux/Dushman method............................ 67
8
Figure 3.8. Micromixer performance and efficiency ....................................................... 69
Figure 4.1. Reaction setup for kinetic study of DHT 2 formation................................... 76
Figure 4.2. Schematic and photograph of Teflon® AF based degasser ........................... 80
Figure 4.3. Reaction setup for kinetic study of the direct NaNT 3 synthesis. ................. 81
Figure 4.4. DHT 2 reaction order determination through consumption of nitride .......... 84
Figure 4.5. Arrhenius correlation between temperature and DHT 2 rate constant. ......... 85
Figure 4.6. NaNT 3 reaction order determination through formation of NaNT 3 ........... 86
Figure 4.7. Reactant concentration dependence on pH at constant T and I..................... 88
Figure 4.8. NaNT 3 generation rate vs. pH and ionic strength ........................................ 90
Figure 4.9. Arrhenius correlation between temperature and NaNT 3 rate constant. ....... 91
Figure 4.10. Conversion to NaNT 3 with flow rate through a fixed volume................... 93
Figure 4.11. Conversion to NaNT 3 vs. temperature at slightly basic pH....................... 94
Figure 5.1. Two-thermal-zone microreactor.................................................................. 103
Figure 5.2. Photograph of assembled microreactor system ........................................... 105
Figure 5.3. Finite element modeling of heat transfer in the reactor setup. .................... 106
Figure 5.4. Cross-section of the temperature profile along the reactor ......................... 107
Figure 5.5. Aminolysis of 22 with 6 in microreactor at 195ºC and 250 psi .................. 114
Figure 5.6. Product distributions of aminolysis of SO 5 with 23 .................................. 116
Figure 5.7. Solvent study of SO 5 aminolysis with 1.2 equiv. of AI 22........................ 117
Figure 5.8. Production of metoprolol 19 at various amine 17 ratios ............................. 119
Figure 5.9. Aminolysis to form indacaterol precursor 15 in a microreactor.................. 121
Figure 5.10. Bisalkylation reaction order determination ............................................... 124
Figure 5.11. Arrhenius correlation of bisalkylation....................................................... 125
Figure 5.12. Aminolysis reaction order determination .................................................. 127
Figure 5.13. Arrhenius correlation of SO 5 aminolysis. ................................................ 128
Figure 5.14. Experimental vs. calculated yields for aminolysis of SO 5 with AI 6. ..... 129
Figure 5.15. Modeled yield of 7 with time .................................................................... 130
Figure 5.16. Calculated residence time distribution in a tube reactor. .......................... 133
Figure 5.17. Cartoon of the effect of Dean flow on fluid mixing.................................. 134
Figure 6.1. Experimental microfluidic system for the solids-generating reaction......... 141
Figure 6.2. Serpentine microreactor used for the initial solids handling tests............... 143
9
Figure 6.3. Microchannel clogging by solids agglomeration ........................................ 144
Figure 6.4. Microchannel clogging by wall deposition ................................................. 145
Figure 6.5. Silicon microreactor with a gradual spiral main channel ............................ 147
Figure 6.6. Wall deposition in a spiral microreactor ..................................................... 147
Figure 6.7. Redesigned spiral microreactor with enlarged channels and quench.......... 148
Figure 6.8. Sequential addition of (a) catalyst and (b) aryl amine................................. 151
Figure 6.9. Sequential addition reactor design as a circuit diagram. ............................. 152
Figure 6.10. Sequential addition spiral microreactor..................................................... 153
Figure 6.11. Sequential addition spiral microreactor with Rhodamine B...................... 155
Figure 6.12. Silicon microreactor before and after PTFE surface coating .................... 156
Figure 6.13. DRIE fluoropolymer passivation coating in a microchannel. ................... 157
Figure 6.14. PTFE-coated microchannel following flow of 20 wt% KOH at 60ºC ...... 158
Figure 6.15. Ultrasonication bath waveforms................................................................ 160
Figure 6.16. Smoothing of the absolute values of the data in Figure 6.15 .................... 161
Figure 6.17. Aluminum chuck for microreactor heating and direct acoustics............... 162
Figure 6.18. Ultrasound influence on product yield for different catalyst loadings. ..... 164
Figure 6.19. RTD for liquid flow with and without ultrasound..................................... 167
Figure 6.20. RTD for solids-containing flow with and without ultrasound................... 167
Figure 6.21. Particle size distribution for Figure 6.18 with and without ultrasound ..... 168
Figure 6.22. Pressure drop vs. time for the C-N coupling in the acousticsirradiated sequential-addition silicon microreactor. ............................................... 169
Figure A.1. Goldilocks reactor mask ..............................................................................195
Figure A.2. KOH-etched meso-reactor masks................................................................196
Figure A.3. HNA-etched meso-reactor mask. ................................................................197
Figure A.4. First-generation micromixer masks .............................................................200
Figure A.5. Second-generation micromixer masks.........................................................201
Figure A.6. Spiral single-addition reactors .....................................................................202
Figure A.7. Spiral sequential-addition reactor................................................................204
10
List of Tables
Table 5.1. Epoxide aminolyses in a microreactor vs. microwave (μw) batch. .............. 113
Table A.1. CORAL Abbreviations .................................................................................193
Table A.2. “Goldilocks” KOH-etched reactors ..............................................................194
Table A.3. HNA-etched mesoreactor..............................................................................196
Table A.4. First-generation micromixer .........................................................................197
Table A.5. Second-generation micromixer.....................................................................200
Table A.6. Spiral sequential-addition reactor .................................................................202
List of Schemes
Scheme 3.1. Competing parallel reactions of the Villermaux/Dushman method............. 66
Scheme 4.1. Formation of NaNT 3 from 5-AT 1 (AT) via DHT 2 intermediate ............ 74
Scheme 5.1. General epoxide aminolysis synthesis......................................................... 99
Scheme 5.2. Expanded epoxide aminolysis reaction scheme with SO 5 and AI 6........ 100
Scheme 5.3. Synthesis of 15 as a precursor to indacaterol 12 ....................................... 101
Scheme 5.4. Synthesis of metoprolol 20 from epoxide 18 and isopropylamine 19....... 102
Scheme 5.5. Epoxides and amines used as model substrates. ....................................... 112
Scheme 5.6. Aminolysis of SO 5 with aniline 23; product 30 is often favored............. 116
Scheme 6.1. Palladium-catalyzed amination of 4-chloroanisole with aniline. .............. 140
Scheme 6.2. Proposed mechanism for the model chemistry. ........................................ 150
11
Chapter 1.
Introduction
1.1
Background – Microchemical Systems
The development of silicon microfabrication techniques for the semiconductor
industry1 has, in turn, enabled other disciplines to develop microscale devices with a high
degree of customizability and precision.
Application of mechanical engineering to
microscale devices has led to the field of microelectromechanical systems, or MEMS.2
Similarly, the combination of chemical engineering principles, integrated with rapid
separation and inline analytical technology, has brought about the development of micrototal analytical systems, or μTAS.3
As a nascent technology, microreactors were developed in the 1990s.3-8 One of the
earlier examples of a silicon microreactor system was developed at DuPont as
interconnected unit operations.9-11 Since then, a great deal of technological advancement
has been made in the field of microfluidic chemical application, both in developing
microreactor techniques and in applying them to improved study and processing of
chemical syntheses, as recently reviewed by Hartman and Jensen.12
A microreactor, by definition, is a device with functional features that are submillimeter in active radius (and often on the order of microns), designed to provide a
platform for chemical synthesis. In addition to chemical synthesis, microdevices such as
separators, pumps, and flow cells can enable microsystems for multistep synthesis and
reaction workup on a continuous scale. A microreactor is typically several centimeters
across and is designed for continuous-flow operation through flow channels that are
fractions of a millimeter in depth. The reactor volume is most commonly defined by a
single long channel up to several meters in length, although parallel-flow reactors with
channel manifolds are also used.
The small flow dimensions of the microreactor allow for very low-Reynolds-number,
high-Péclet-number flow profiles, making diffusion and dispersion very easy to
12
characterize and control. In addition, fabricating microreactors out of silicon, which has
a high thermal conductivity of 148 W/m·K,13 allows for very fast thermal equilibration
and highly precise temperature control.
Combined, these features make silicon
microreactors unrivaled study tools for reaction screening and kinetics.
In addition to precise reaction control, microreactors have proven to be much safer
than large-scale flow and batch vessels. The high ratio of surface area to volume in
microreactors provides enhanced heat and mass transfer, useful in such cases as the
highly exothermal (-473 kJ/mol) selective direct fluorination of toluene14 and in
suppressing flame formation by quenching free-radical generation in hydrogen peroxide
synthesis.7
Microreactors have been applied for multiphase reaction acceleration,15 as well as for
on-demand synthesis of hazardous intermediates,16 including conversion of chlorine to
phosgene.17 Thus, their utility has been demonstrated on the microscale. By developing
techniques and methods for rapid and efficient reaction evaluation and kinetic study,
microreactor systems can be applied to industrial synthetic schemes and can greatly
improve overall process design and development.
1.2
Motivation
In the fine chemical industries, including pharmaceuticals, cosmetics, food additives,
agricultural materials, and many other fields, a great deal of time, effort, and cost is
dedicated to the development of synthesis processes. Large amounts of reagents and
energy are consumed to take a process developed by the chemist in small flasks and to
produce a full-scale, multistep, integrated pathway for commercial production.
Additionally, many chemical reactions include unstable or difficult-to-isolate
intermediates, the production of which is not straightforward to optimize due to their
transient nature. Numerous reactions are hazardous to perform, either due to aggressive
conditions, toxic reagents, or explosive or flammable intermediates or products.
Reducing the scale of such reactions, even in batch, can reduce the hazards to the
researcher,18 minimize the time and material consumption for such studies, and provide
insight into physical and chemical processes to obtain greater understanding of the
systems.
13
The process of optimizing a reaction can be tedious, expensive, and protracted. In the
cases of catalytic reactions, a typical lab may be able to perform only tens of experiments
for catalyst and ligand screenings, while an industrial combinatorial chemistry system
may be used to run hundreds, if not thousands, of reaction experiments to be performed,
each consuming a quantity of typically costly organometallic materials.
Once a
satisfactory catalyst combination is selected, it is necessary to optimize the reaction
conditions to determine a set of parameters such as temperature, stoichiometry,
concentration, residence time, etc. at which the most economically beneficial result is
obtained. Again, this gamut of parameters can require a large number of experiments.
Performing these processes in a microreactor system can greatly increase the efficiency
of this process.
The ability to manipulate microliter volumes can allow for rapid
sequential screenings, consuming only micromole quantities of reagents for each
experiment.19 Moreover, the ability to precisely control temperatures and flow profiles
ensures that the obtained kinetics are accurate and applicable to a broad range of systems.
The ability to perform reactions in flow allows for rapid changes in conditions and
reagent proportions with little effort, allowing for a wide range of reaction parameters to
be analyzed with a single set of reagent preparations.
Many chemical processes are difficult to perform well by conventional methods such
as in batch reactors or in macro-scale flow. Reactions that have long residence times are
time-consuming to study and optimize. Syntheses that involve solids cannot be flowed
easily, and there are often mass transport limitations that hinder full understanding of the
chemistry. Performing chemistries at high temperatures and pressures renders them
difficult, and possibly hazardous, to sample during operation. Chemical pathways that
require the synthesis of hazardous intermediates are inherently dangerous to operate. The
application of microreactor systems can address many of these challenges, providing the
ability to safely perform and sample chemistries at a broad range of conditions, to
precisely evaluate reaction kinetics, and to safely and efficiently synthesize hazardous
intermediates. There is strong industrial interest in continuous processing, both for study
and production.20 With a toolbox of microfluidics, many industrial process research and
development challenges can be met.
14
1.3
Thesis Objectives and Outline
To support large research organizations, as well as small academic laboratories, a
method of rapidly and inexpensively producing a wide range of silicon microdevices is
necessary.
Typical fabrication schemes, while highly robust and flexible regarding
possible reactor layouts, are limited to a one-wafer-at-a-time etch technique that, in
addition to being very time-consuming, is also quite expensive for many research
initiatives. To overcome this bottleneck in processing and to reduce the fabrication cost
of devices, wet-etch-based alternatives have been developed. Chapter 2 describes the
process development and reactor design using two different wet etch techniques to
rapidly mass-fabricate batches of silicon reactors.
Many reaction chemistries contain extremely rapid steps, which are often not well
understood regarding their kinetics because of mass transfer limitations. For reactions
that are highly sensitive to changes in pH or to reagent concentrations, the elimination of
concentration gradients at a rate several orders of magnitude faster than the kinetics to be
studied is necessary. Additionally, fast mixing remains necessary at large flow scales, as
well, requiring a device capable of eliminating concentration gradients without large
energy expenditure. To that end, a rapid liquid-flow micromixer with a low pressure
drop is designed and developed, as discussed in Chapter 3.
The application of fast microscale mixing to precisely and safely study the kinetics of
a rapid reaction with a highly energetic intermediate is demonstrated in Chapter 4. The
multi-step synthesis of a nitrotetrazolate compound via a diazonium intermediate in a
gas-generating reaction is studied, and the kinetics of both reaction steps are evaluated
with accuracy. The synthesis of this energetic compound is too hazardous to characterize
through batch experiments; however, using microscale flow chemistry, both the kinetic
parameters of each reaction step and the equilibrium of the reactive intermediate are
safely determined. Additionally, a slightly modified setup using the same micromixers is
applied, based on the determined kinetic parameters, to a scale-up of this reaction, safely
generating production-level amounts of the nitrotetrazolate compound with a small lab
bench footprint.
To demonstrate the capability of microreactor-enabled conditions to accelerate
chemical reactions, Chapter 5 relates the study of β-alcohol formation by epoxide
15
aminolysis. Through the use of microreactors, the reactions are able to be performed at
high pressures, reaching high temperatures in liquid phase while using volatile solvents
and reagents.
Reactions are shown to be greatly accelerated from their traditional
methods, with pharmaceutically relevant compounds synthesized at residence times
decreased by a factor of 30-60. A set of model chemistries is explored, providing
fundamental understanding of the effects of parameters such as solvent and co-solvent
effects and steric hindrance.
Additionally, the high utility of the microreactors is
demonstrated by enabling a full kinetic study of a multistep mechanism within a short
time and with only grams of reagents.
Reactions that produce solids as products or byproducts are highly difficult to
perform in flow and thus to efficiently study on the microscale. Chapter 6 describes the
use of silicon microreactors to gain fundamental understanding of flow stoppage by
solids in microchannels. A palladium-catalyzed C-N coupling reaction between an aryl
amine and a substituted aryl halide is used as the model chemistry. Several techniques
are evaluated to learn how solid-containing slurries can be made flowable in
microchannels. A combination of reactor design and application of acoustic irradiation
enabled the flow of the reaction with moderate yield.
This has high potential for
expanding the microfluidic toolbox to the vast number of chemical syntheses that feature
solids, thus allowing them to be studied in microscale systems to attain the
aforementioned advantages of reaction acceleration, precise kinetics, and safe, rapid, and
efficient reaction study.
Finally, Chapter 7 provides some concluding remarks and
outlook of future developments in the application of microfluidics to the study of
industrially interesting but difficult syntheses.
16
Chapter 2.
Wet-Etch Fabrication
Silicon microfabrication can be expensive and highly time-consuming. However, for
scale-out (parallelization) of silicon fluidic systems, a large number of identical reactors
may be necessary for efficient production processes. Similarly, for producing siliconbased full-wafer devices, the ability to simultaneously perform the etching step of the
fabrication on multiple wafers would be invaluable for reducing the fabrication cost and
time requirement. Acid and base wet-etch techniques were developed and implemented
for successful production of silicon micro- and meso-reactors.
A potassium hydroxide etching method was designed to take advantage of silicon
crystal properties, producing features etched to multiple depths, including through-holes,
in a single etch step.
This single-etch-step method was used to produce a set of
microreactors possessing channels with both rectangular and triangular cross-sections of
multiple depths.
Additionally, up to 13 wafers were able to be reliably processed
simultaneously.
An HNA (hydrofluoric/nitric/acetic acids) etching method was designed for the
production of robust full-wafer mesoscale reactors. HNA can be extremely rapid (600μm-deep etch in 10 minutes) and etches all crystal planes at equal rates, producing
features that are nearly semicircular in cross-section. This process was used to fabricate a
mesoscale reactor for reaction scale-up studies, consisting of a single channel 1.25 mm
wide and 0.6 mm deep, providing a volume of 5 mL in a compact, easy-to-handle device.
17
2.1
Silicon Microreactor Etching
At its most basic level, a flow reactor is a sealed vessel (often channel-shaped) with
one or more inlets and outlets. Thus, there are several general requirements for the
fabrication of any silicon-based fluidic device. One must be able to remove some amount
of silicon to create the vessel (channel) shape, to seal said vessel, and to provide fluid
access to it, not necessarily in that order. The sealing is typically done by bonding the
processed silicon wafer to a capping wafer, either silicon (through fusion bonding)21 or a
borosilicate glass (through anodic bonding).22 In either case, the capping wafer can also
have been processed, thus having reactor features on both wafers (or even to form 3dimensional structures from stacks of more than two patterned wafers).23 The fluidic
access can be provided either as part of the reactor features, such as open-ended
channels,24,
25
produced in a separate etch step similar to main device etching,15,
26
or
micromachined in the capping wafer (in the case of a glass capping wafer, this may be
done by laser ablation, ultrasonic drilling, or milling).23 Finally, but possibly most
importantly, the reactor features can be etched by a variety of methods, including plasma
and wet etch techniques.2
Silicon VLSI (very-large-scale integration) technology allows for a wide range of
additional elaborations on this basic design, such as surface modification by oxide
growth, chemical vapor deposition, and evaporated metal deposition,27 creating improved
chemical resistance properties,28, 29 integrated resistive heaters and thermocouples,30 and
bond pads for solder-based fluidic connections.31 However, even with these elaborate
components, the silicon etch step to create the channels and the flow ports is often the
most costly, at times accounting for the majority of the reactor processing cost.32 This
cost, though, strongly depends on the choice of the etching technique, with ones that are
the easiest to apply in terms of design flexibility, process automation, and ease of use also
being the most costly and time-consuming. The different etching techniques have been
reviewed and compared in literature33 and in various textbooks on silicon fabrication.1, 2,
27
Each has its advantages and disadvantages; however, to date, there have been no
literature reports of a cheap and efficient technique applied to produce flexible, deepfeature designs in silicon fluidic devices.
18
2.1.1 Plasma Etching
One of the most preferred methods for creating deep features in silicon is through a
plasma etch technique called deep reactive ion etching (DRIE), a variant of reactive ion
etching (RIE).2 Plasma etching acts by creating reactive ions that are able to remove
exposed silicon atoms into the vapor phase.34 In RIE, the reactive plasma is formed by
charged SF6, and the high charge below the substrate causes the ionized gas molecules to
impact the silicon substrate nearly normal to the surface. This leads to a high degree of
anisotropy for etches of short duration, although significant undercutting of the mask will
occur after prolonged etch times and depths. Thus, this is a very useful method in the
semiconductor industry for the fabrication of small steps or vias in the silicon substrate.
To expand the utility of RIE, researchers at Bosch developed the DRIE variant.35 For
many chemical species, plasma conditions lead to their polymerization. DRIE takes
advantage of that effect by alternating the RIE etching step with one that introduces C4F8
plasma into the chamber. In that step, the substrate charge is reduced; thus, ions are not
as strongly drawn downward, and the polymerizing fluorocarbon deposits evenly on all
exposed wafer surfaces. When the subsequent plasma etch step occurs, the prevalent
downward-directed ions rapidly remove the fluoropolymer layer on the bottoms of the
features and continue to etch the silicon substrate, while the side walls are protected from
the occasional angled ions. Alternating between the C4F8 passivation and the SF6 etching
allows the etching of arbitrarily deep features with very high aspect ratios. Figure 2.1
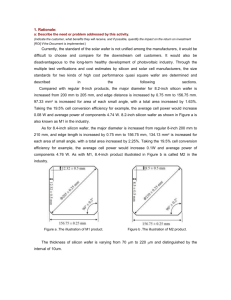
shows the etch profiles created by RIE and DRIE.
(a)
Silicon
(b)
Photoresist
Figure 2.1. Etch profiles formed by RIE (a) and DRIE (b).
A number of advantages make DRIE highly attractive. First, as mentioned above, it
is possible to etch to arbitrary depths with nearly vertical side walls. This feature allows
for the production of a wide variety of designs, as any two-dimensional feature or
drawing will be extruded vertically into the depth of the silicon wafer. Device design
thus becomes limited only by the resolution of lithography in the masking layer. The
19
second advantage is a relatively high selectivity of the etchant for silicon over certain
photoresists (40:1 etch rate ratios), allowing for a simple and relatively inexpensive
method of producing mask layers and defining features. Similarly, DRIE has a high
selectivity for silicon over thermal silicon oxide (100:1 etch rate ratio), making the
frequently used thin layers of thermal oxide a good functional mask for DRIE, especially
if nested mask use is desired. Finally, the high degree of control that modern DRIE
equipment allows over the gas composition, plasma power, chamber pressure, and
process timing allows for tailoring of etch methods for different applications, as well as a
high degree of reproducibility for the same method.
On the other hand, there are several disadvantages to DRIE that may lead one to
consider other etching methods. First, because DRIE is in fact not truly anisotropic, but
is rather a series of slightly isotropic etches interspersed with passivation steps, the side
walls are not smooth. Instead, they are scalloped, as shown in Figure 2.2a, creating
micron-level surface texture. Additionally, the etch creates a high degree of sub-micron
surface roughness due to the incident ion bombardment. Therefore, without an additional
HNA polishing step (see section 2.1.3 for more details), the process results in submicroscopically rough side walls, as shown in the scanning electron microscopy image of
Figure 2.2b. Second, to produce through-holes, additional steps are necessary to mount
the substrate wafer onto a handle to prevent the helium coolant flow from leaking into the
vacuum chamber, thus requiring additional processing time.
(a)
(b)
Figure 2.2. Scanning electron microscopy images of DRIE-etched side wall, showing (a) scalloping36
and (b) surface roughness.
With DRIE, the etch depth uniformity across the wafer surface is strongly dependent
on the etch parameters, with there being a trade-off between etch rate, selectivity,
20
anisotropy, and uniformity.37 The etch recipes must select a middle ground to have
reasonable values for all of these properties. Thus, the methods preferred by users
experienced on the particular piece of DRIE equipment available to us typically have
high anisotropy and etch selectivity, a reasonable etch rate of 1 to 2 μm/min, and a depth
variation of approx. 10% from wafer center to wafer edge.
This is a significant
difference that often requires reactor designs, layouts, and arrangements on the wafer to
have depth-insensitive features (such as through-holes or halo etches) nearer to the edge
of the wafer to allow more control over the depth of more sensitive features.
Additionally, the aforementioned etch rate means that for thicker wafers of 1000 μm that
require through-holes, it is not uncommon to require 12 to 15 hours of combined etch
time per wafer. When combined with the fact that DRIE can only be performed on a
single wafer at a time, it is easy to see how the etch step can become a significant
bottleneck when it is necessary to process several wafers, especially if full-wafer devices
or a multi-wafer stack are desired.
DRIE requires a high degree of technology to control the process;38 combined with
the cost of the reactive gas and the high energy expenditure over very long periods of
time, this makes DRIE one of the most expensive microfabrication processes per wafer32
when deep features and/or through-holes are required by the process.
All of the
discussed shortcomings are the reason for developing alternative etch processes to be
inexpensive, robust, and reproducible.
2.1.2 Potassium Hydroxide Etching
There are several well-known wet etch methods for silicon. Methods that use caustic
aqueous solutions, such as potassium hydroxide (KOH) and tetramethylammonium
hydroxide (TMAH), the two most widely used etchants, act via the hydroxide ions, which
are thought to react with silicon to form silicon hydroxide anions, as well as hydrogen
gas. Caustic wet etches have high selectivities towards certain crystal planes, with etch
rate ratios of as high as 100:1 for {100} vs. {111} crystal planes.39 This crystal plane
selectivity lets certain planes act as etch stops, resulting in constrained, regular features.
Although an aqueous solution of KOH can be used as the etchant to anisotropically
etch vertical features in (110)-oriented silicon wafers,40 such wafers are typically very
21
expensive; thus, (100)-oriented wafers were our substrate of choice. Applying KOH to
(100)-oriented silicon wafers (respectively, the most commonly available, and thus the
least costly, etchant and silicon wafers), the etch stop planes are at a 54.74º angle to the
surface.41 Thus, any feature, if etched sufficiently deeply, will form either a pyramidal or
a triangular-prism pit bounded by {111} crystal planes (Figure 2.3). This feature can be
both an advantage and a hindrance. It is advantageous because it ensures that all etched
features are aligned to crystal planes, which is necessary for devices such as
waveguides23 and optics.42, 43 However, it also means that any feature that is misaligned
from the crystal plane in lithography will etch out larger than is designed, as the bounding
etch planes will be slightly outside the feature bounds.
(a)
Silicon
Nitride
(b)
Figure 2.3. Illustrations of features etched by KOH: side view (a) and top view (b); the dashed line
represents the cut along which the side view is made.
For the etching procedure of aqueous KOH on (100)-oriented silicon wafers, design
of features is highly important, especially for fluidic devices such as microreactors.
Nearly all features to be etched by KOH are designed to be rectangular and aligned along
the <110> direction. These features form either pyramidal pits or long straight channels
with triangular or trapezoidal cross-sections. These have been used in the past for
parallel-channel reactors23 and fiber alignment channels.31 However, knowing the etch
rates of the crystal planes, it has been possible to develop methods to compensate for
convex corner etching.44-46 These methods have been used to create series of channels
that take 90º turns (always aligned along the <110> direction), thus expanding the
possible length of the fluidic channels that can be used for microreactors.
There are several caveats, however, to using these corner compensation methods.
First, the corner compensation features must be calculated for a specific etch depth.
Therefore, each design and mask is only usable for reactors of one depth. A related issue
is that the desired etch depth determines the size of the corner compensation features.
22
Thus, deeper etches require larger corner compensation features, limiting how closely
spaced adjacent channels can be placed and increasing unutilized silicon area. If multiple
features with convex corners are desired to have different depths, then the all of the
corner compensation features must be sized to the maximum desired depth. Otherwise,
the shallower features will have the convex corners be etched inward and deeper due to
the unavailability of the etch-terminating planes (this, however, may be acceptable for
certain features such as low-volume mixing zones).
Nitride
(a)
Silicon
(b)
Figure 2.4. Illustration of side view (a) and top view (b) of a 90º turn aligned to <100> direction; the
dashed line represents the cut along which the side view is made.
(a)
(b)
Figure 2.5. Photographs of the inside corner of a 90º turn and the outside corners of a 180º turn with
KOH etching (the etched area is silver-grey, and the unetched area is dark-green).
Another method of etching channel-type features with KOH in (100) wafers is by
aligning the long rectangular features along the <100> direction.47 This places the etchterminating {111} planes 45º to the feature sides, and the fast-etching {100} planes are
now parallel to the features. As a result, the feature etches both downward and laterally,
undercutting the mask along the long sides of the rectangular feature at the same rate as
the vertical etch (Figure 2.4). In the process, channels with perfectly vertical walls and a
sharply delineated rectangular cross-section are formed along most of the feature. At the
short sides of the rectangle features, the channel is bounded by the {111} planes aligned
45º to the original feature – the <110> direction – at the feature corners, sloping into the
23
etch at a 54.74º angle. Thus, if two long rectangular channels are joined at a 90º angle,
the turn would have a perfectly sharp 90º corner with vertical side walls on the inside
turn, and two vertical walls connected by a 54.74º-sloping wall at two 135º angles on the
outside turn (Figure 2.5).
Using the method of channel alignment to the <100> direction can be very useful, as
channels can now be placed arbitrarily close to each other, providing better utilization of
silicon area. It is important that lithographic features to produce these channels be
designed accounting for the lateral etch, the rate of which will now be equal to the
vertical etch.
Another major advantage is that this method produces rectangular
channels, which have significantly less pressure drop for the same volume at the same
etch depth.
Additionally, the rectangular cross-section is more familiar to those
experienced with DRIE or bulk machining for microreactor production.
KOH etching has several advantages over processes such as DRIE.
One such
advantage of this process is the highly smooth walls formed by the crystalline etch.
Secondly, features with multiple depths can be achieved in a single etch step. By
aligning certain features along the <110> direction and allowing them to self-terminate
while having other features aligned along the <100> direction, it is possible, for example,
to create a reactor with relatively deep channels with rectangular cross-sections, with
shallow triangular channels for a mixing zone, with less shallow triangular channels for a
quench mixer, and having very shallow alignment marks on the wafer (here, the
shallowness is desirable to reduce undue stress on the wafer from unnecessary etches).
Considerations must be made, though, for corner compensation limitations for the
triangular channels with large depth disparity. Additionally, because the wafer would be
exposed to the KOH solution equally on both sides, it is possible to simultaneously etch
features on both sides of the wafer. Thus, through-hole ports and, for example, channels
for heat exchanger fluid can be placed on the back side at the same time. This also
reduces the necessary etch time as compared to DRIE. At typical process conditions, the
etch rate of the KOH process is similar to that DRIE, 1 to 2 μm/min. However, because
through-holes are etched at the same time, a process requiring 700-μm-deep channels
with ports in a 1000-μm-thick wafer requires only 70% of the etch time of DRIE for the
same features.
24
A predominant advantage of KOH etching is its high uniformity. The KOH etch is
kinetics-limited and not limited by diffusion. With a sufficiently good temperature bath
to ensure no thermal gradients across the wafer and wafer-to-wafer (a fairly easy task),
the etch difference across the wafer and between wafers is often as low as 1%. As many
as 25 wafers can be easily placed in a single wafer boat and etch container. Thus, 2 sides
each of 25 wafers can be uniformly processed with a single etch step, or 50 times the
throughput of DRIE. Combined with the extremely low cost of KOH, the low energy
expenditure, and the low level of technology required for this process, this renders KOH
etching a far cheaper alternative to DRIE.
There are several disadvantages to using KOH, as well. First, there is far less
flexibility in reactor design as compared to DRIE. Because one is limited by the crystal
planes, features must be aligned along the <110> or <100> directions on the wafer, and
only 45º and 90º turns can be formed, with no possibility for curvature. Moreover, the
features aligned in the <100> direction will always have a width of twice the depth plus
the lithographic feature width. Thus, the minimum aspect ratio of those features can only
approach (but never reach) 2. Second, the mask for the etch must be either silicon oxide
(suitable for relatively shallow etches) or silicon nitride (necessary for deeper etches).
Both cases require additional processing steps, and for silicon nitride, not an insignificant
additional cost. However, the etch rate of silicon nitride in KOH is very low, and a 0.2μm film of nitride is sufficient for a 750-μm-deep etch. Additionally, up to 50 wafers can
be processed at once for silicon nitride deposition. Thus, a thin layer of nitride and a
large batch size can make the cost per wafer very low.
Another disadvantage of KOH is the high sensitivity of the etch to the quality of the
lithography, especially for features aligned in the <100> direction, where the lithographic
features are much finer than the desired etched ones to account for the lateral etch. In
those cases, very fine breaks in the feature would cause channel terminuses because they
would be immediately bounded by etch-stop planes. Similarly, slight enlargements of the
features may cause breaks in channel walls due to the lateral etch if thin walls are desired
(see section 2.3.1 for discussion and illustrations). With <100>-aligned features, etch
stops such as a buried oxide layer would not stop the lateral etch, reducing the utility of
that method. Finally, because the etches are along crystal planes, especially for channels
25
with rectangular cross-sections, there are high stress concentrations along the etched
corners, lying directly on crystal planes, thus making KOH-etched wafers much more
fragile and resulting reactors less robust to stress (see section 2.3.3 for discussion and
illustrations). However, a brief HNA etch step following the KOH process can smoothen
the crystal corners and disperse the stress accumulation.
2.1.3 Acidic Wet Etching
In addition to the caustic wet etching methods described in the previous subsection,
acidic wet etch methods also exist. The best-known such method is one that uses a
mixture of hydrofluoric, nitric, and acetic acids (HNA).48 This etch solution has been
well characterized in terms of the silicon etch rates in mixtures with varying proportions
of the three components.49 The nitric acid component is a strong oxidant, capable of
acting on silicon to form silicon oxide. The second active component, hydrofluoric acid,
is an etchant of silicon oxide; thus, in HNA, it removes the SiO2 formed by nitric acid
oxidation, exposing more bare silicon. Between the two acids, silicon becomes etched
isotropically, forming quarter-circular undercuts under the exposed feature edges. The
purpose of acetic acid in the mixture is only as a dilutant; while water can be used, it also
changes the dissociations of HF andHNO3, thus making an acid preferable for this
purpose. A diluent is necessary to be able to control this process, which is otherwise
extremely rapid and highly exothermal.
As briefly mentioned above, HNA is commonly used for various polishing and
crystal plane stress removal purposes. Because it is a fully isotropic process (assuming
sufficient mass transfer), etching occurs fastest around the greatest surface area; thus,
corners formed by KOH and micron-scale ridges, bumps, and protrusions that are a
product of DRIE can be quickly removed by fairly mild HNA. However, HNA is a
highly aggressive etchant, and no masking technique is very resilient to it. For that
reason, to the best of my knowledge, it has not been used for deep features.
The strongly oxidative property of nitric acid makes the selection of a mask for deep
etching rather difficult. Organic masking layers such as photoresist are removed by HNA
in a matter of seconds. Similarly, most metals are rapidly oxidized by nitric acid, not to
mention the large cost associated with metal deposition. It is physically feasible to use
26
electron-beam evaporation to deposit a thin layer of gold atop an adhesion layer of
titanium, as gold would easily withstand HNA; however, the titanium oxide/silicon oxide
forming the true adhesion layer will likely succumb to the etchant after a short period.
Additionally, the cost of the metals and the use of equipment, combined with the small
number of wafers capable of being processed simultaneously (4 on the machine available
to us) make this application prohibitively expensive. As hydrofluoric acid etches silicon
oxide, that material is not usable as a masking layer.
The material typically used as a mask for HNA is silicon nitride deposited by lowpressure chemical vapor deposition (LP-CVD). It has excellent adhesion properties to
silicon, without peeling even at the most aggressive conditions. Additionally, it is fairly
resilient to HNA, with HNA selectivity toward silicon versus silicon nitride of as high as
600:1, depending on the temperature and HNA composition. However, it is impractical
to deposit silicon nitride with large thicknesses; 2 μm is the maximum deposition
thickness in a single coating layer.
This is not a major limitation because, if an
appropriate composition of HNA is used, this is sufficient to allow etching down to 650700 μm, producing quite deep features for large fluidic devices. Moreover, silicon nitride
deposition can be a moderately expensive process per batch,32 although this is moderated
by the ability to coat up to 50 wafers in a single batch.
As with any other technique, HNA offers a number of advantages and possesses
several disadvantages as compared to KOH and DRIE. Being a wet-etch technique
similar to KOH, HNA enables the simultaneous etching of multiple wafers in a single
batch.
It also allows simultaneous etching of features on both sides of the wafer;
however, with HNA, it is not possible to design an etch stop. Thus, two-sided processing
can be used to produce through-holes or to create sets of features that are both less than
half of the wafer thickness; it is not possible to etch to multiple depths in a single step.
HNA produces features that are rounded in cross-section. With sufficiently fine
lithographic features, the cross-sections of channels would be nearly semicircular,
although this depends on the mass transfer (Figure 2.6). This may be advantageous for
applications requiring lower dispersion, especially multiphase liquid or gas-liquid flows,
where having two corners instead of four would reduce the dispersal of the wetted phase
under proper flow conditions. Additionally, such a wafer can be fusion-bonded to one
27
that has a mirror-image pattern, forming a sealed silicon channel with a nearly circular
cross-section and thus reproducing the familiar polymer or steel tube, but with the
advantageous properties of silicon and with far more freedom of reactor layout.
Although, similarly to KOH, any HNA-etched feature would have an aspect ratio of
greater than 2, the lack of crystal plane limitation significantly expands the flexibility of
reactor design, allowing for arbitrary shapes such as winds, serpentines, and spirals.
Moreover, because the etch is not along any crystal planes and because no sharp corners
are formed within the silicon, there are no places of high stress accumulation.
In
addition, the rounded features ensure that the force of the pressure from the liquid is
normal to the bulk of the silicon and is dispersed in all directions and not focused on any
one plane.
Thus, the HNA-etched reactors are expected to be the most robust to
pressures and stresses for the same feature sizes and layouts.
Nitride
(a)
Silicon
(b)
Figure 2.6. Illustrations of HNA-etch profiles with (a) and without (b) agitation.33
Another feature of HNA etching is its extremely fast etch rate under certain
compositions, which is both an advantage and a hindrance.
Because HNA etch is
exothermal, it can also be difficult to control at the faster etch rates due to the inability to
sufficiently rapidly remove heat.
At slower etch rate compositions, however, the
selectivity toward silicon over nitride also significantly decreases. Nonetheless, at the
composition found to be optimal for both etch rate and selectivity (see section 2.3.3 for a
detailed discussion), the etch rate of silicon was 100 μm/min, or 50-100 times faster than
either KOH or DRIE. The fast etch rate, however, makes this etch somewhat masstransfer-limited. Thus, a very good mixing strategy is necessary to ensure uniform
etching across the wafer and wafer-to-wafer. An etch difference of ~10% was typically
found between the center and the edge of the wafer; however, this being an isotropic etch,
this causes a cross-sectional area difference of ~20%, making an etch rate difference
similar to that with DRIE create a much more significant channel volume difference.
28
The fast etch rate makes this process ideal for fabrication of large devices such as
full-wafer fluidic reactors, where one wants to process many wafers at once. Even
without the availability of good mixing to ensure good wafer-to-wafer uniformity within
the tank, a magnetic stirrer is sufficient to provide acceptable across-wafer uniformity.
With a fast etch process, it is easy to rapidly process many wafers sequentially using only
a small amount of material, making this a highly economical process.
2.2
Reactor Design and Fabrication
Fluidic reactors were designed with consideration for the most efficient fabrication
route within the constraints of the available microfabrication facility. Additionally, it was
desirable to maximize the utility of the silicon surface; i.e., the designs were made to
produce the maximum total reactor volume from a single silicon wafer. A design was
made for a set of microreactors to utilize the etch-controlling advantages of KOH etching.
To produce mesoscale reactors, separate designs were made for both KOH and HNA
etching techniques. In both cases, single processed silicon wafers were capped with clear
borosilicate wafers to retain visual access to fluid flow and possible reactor fail modes.
2.2.1 KOH-Etched Reactors
Microreactor Design
To take advantage of the ability to etch multiple depths in a single etch using KOH
wet etch processing, it was decided to design a set of reactors that will have shallow, selfterminated channels for rapid mixing of incoming reagents, deep rectangular channels for
the main residence time and for the quench, and through-hole ports for reagent and
quench inlets and the outlet, as well as deposited metal pads for solder-based packaging.
The most frequently used silicon wafer thickness for microreactor fabrication has
been 650 μm; thus, this was the thickness selected for these reactors, as well. It was
decided to mimic the layout of previously designed and widely used DRIE-etched
microreactors15, 19, 31 regarding reactor layout, port positioning, and main channel depth.
It was decided to use only two inlets, as that is the most commonly used configuration. If
more inlet streams are required, it is possible to use a union upstream of the inlets or, if
29
rapid mixing is necessary, a micromixer (for more on those, see Chapter 3). Based on
prior simulations that best quenching occurs using a large volume of quench flow, the
channel following the inlet quench was designed to be quite large, 1.5-mm-wide, to
accommodate such diluent flows.
Based on the aforementioned DRIE design, the channels were designed to be 400-μm
deep, which satisfied the constraint that they be more than half the thickness of the wafer
to allow the simultaneous etch of through-holes. Combined with the lithographical
feature size, the selected depth also set the channel width, as the alignment to produce
vertical side walls also etches silicon in the lateral direction. Based on the quality of the
high-resolution transparency printer (Pageworks, Cambridge, MA), channel lithographic
features of 50-μm width were printed. Thus, the reaction channel were 850 μm in width
(400-μm etch in each direction plus 50 μm of the printed feature). The channel turns
were designed to be even wider (average turn radius of 1.25 mm) to allow reactions with
small amounts of precipitate to maintain flow, as it was previously observed that
precipitates tend to agglomerate and clog around turns. Channels were spaced so as to
allow 100 μm of wall width between them, resulting in a dense channel packing and high
silicon utilization.
For the mixing zone, the channels were aligned along the <110> direction to selfterminate, thus forming shallow channels with a triangular cross-section. Because the
channels had 90º turns, corner compensation was necessary to reduce overetching of the
convex corners (Figure 2.7). However, as the main etch depth was significantly greater
than that of the mixing zone, sizable corner overetching was expected to occur regardless
of the compensation features. This was acceptable for the mixing zone, as it reduced the
pressure drop in that section without significantly affecting mixing effectiveness.
Figure 2.8 shows a comparison of the mask features, intended etched reactor, and an
actual photographed reactor. The black region on the diagram indicates the printed mask
features, whereas the blue region indicates the intended etched reactor area (not including
the convex corner overetch). The orange circles represent the bonding pads of deposited
metal, which would appear on the back side of the reactor. It can be seen that the
fabricated reactor matches the design very well, although, as the expanded section shows,
30
the corner compensations are insufficient for such a disparity between mixing channel
depth and maximum depth.
Figure 2.7. Corner compensation features; the white area represents unetched silicon, black shading
represents the mask design, and blue shading represents the intended etched feature; channel width
is 200 μm, and the finest feature size is 10 μm.
(b)
(a)
Figure 2.8. KOH-etched microreactor: (a) illustration of the mask (black) and the desired etched
features (blue), with bonding metal pads on the back side (orange); (b) photograph of reactor.
Different reaction chemistries may demand different residence times, and because
certain sample volumes are required for analysis, slower reactions often require larger
reactor volumes so as to be able to produce sufficient quantities within a reasonable time.
To that end, three different microreactors were designed for different flow rate ranges,
allowing future users to select from among small (92 μL), medium (200 μL), or large
31
(460 μL) reactors, henceforth referred to as the Goldilocks set.
The volume was
generated by the number and length of the channels, keeping the same depth and width,
thus simulating a user selecting different total lengths of tubing of the same type. Figure
shows the wafer layout with the three designs.
Figure 2.9. Illustration of the layout of the Goldilocks devices on a wafer.
The different reactor designs had different areas for the mixing channels. However,
with simple single-channel laminar mixers, there is a trade-off between pressure drop and
mixing time. Smaller-width channels are desirable for their ability to effect mixing more
rapidly, as the characteristic mixing time is dictated by the diffusion distance, as follows:
t=
x2
2D
2.1
where D is the diffusivity and x is the mixing distance. In the case of triangular channels
and two incoming streams, x is the equivalent diameter, determined by solving the
Navier-Stokes equation for a Newtonian fluid in an isosceles triangle channel. Longer
channels also provide more time for mixing at a given flow rate.
32
Both smaller and longer channels, however, result in greater pressure drop. For
laminar flow through channels, the Hagen-Poiseuille equation dictates the pressure drop,
which is once again dependent on the equivalent radius of the channel. For isosceles
triangles, the Navier-Stokes equation has been solved to give the equivalent radius as a
function of the triangle base (channel width) and angle.50
Thus, for KOH-etched
channels with 54.74º angles, the Hagen-Poiseuille equation can be reduced to the
following:
ΔP = 278
Qμ L
w4
2.2
where ΔP is the pressure drop, Q is the flow rate, μ is the fluid viscosity, L is the channel
length, and w is the maximal (top) width. Thus, it is obvious that pressure drop and
mixing both depend linearly on channel length; however, while mixing time decreases
with the square of channel width, pressure drop increases with the fourth power of
channel width. Therefore, one should maximize the channel width where possible to
prevent an unreasonable pressure drop.
As the smallest reactor was the most constrained in terms of area, it was used as the
basis of the mixing zone design.
Based on the reactor volumes and on previous
experience with chemistry in microscale flow, total reagent flow rates on the order of tens
of microliters per minute were considered typical. Thus, the mixing zone was designed
to provide three times the characteristic mixing time (fully diffused flow to three standard
deviations) at a flow rate of 30 μm/min. The <110>-aligned channels must be spaced
apart at a distance of at least twice the channel width to allow for corner compensation;
otherwise, corner overetch would significantly reduce the mixing channel effectiveness.
With the channel width constraining how many channels can be packed in a given area,
thus limiting the total length of the zone, the maximum width to still allow the minimum
desired mixing was determined at 200 μm. This results in a very tolerable pressure drop
of 5×10-2 bar at 30 μL/min flow of water.
For the larger reactors, the mixing zones were resized to fit within the larger available
areas. As the larger reactors were made so to allow for longer residence times at similar
flow rates, the upper bound of 30 μL/min was maintained. The mixing zones were then
enlarged so as to continue providing three characteristic mixing times at this flow rate,
33
but with a minimal pressure drop. For the medium and large reactors, the mixing channel
widths were 250 and 300 μm, respectively, with channels being spaced farther apart than
twice the channel width. In both cases, the pressure drops were significantly smaller.
Additionally, the greater distance between the channels allowed for larger corner
compensation features. Combined with the larger mixing channel depth (and therefore a
smaller difference between the mixing and reaction channels), this would produce much
smaller corner overetching (see section 2.3.1 for the results).
Mesoscale reactor design
For scale-up of silicon fluidic systems, it is appropriate to investigate scaling effects
in silicon mesoscale devices.
Such devices would afford the advantages of silicon
reactors, such as robustness to high temperatures and pressures, very fast thermal
equilibration, reduction of hot spots due to silicon’s high thermal conductivity, and the
ability too observe the reaction in progress, which is invaluable when studying
multiphase or solids-generating reactions, especially with regard to scaling studies.
(a)
1.55 mm x 0.75 mm channels
(b)
10.5 cm
10.5 cm
Figure 2.10. KOH-etched meso-reactor: (a) illustration of the desired etched features; (b)
photograph of reactor.
The KOH-etching technique was applied to maximize the possible volume etched out
of a silicon wafer. To that end, a 1000-μm-thick wafer was used as the substrate for the
design. It was decided to produce a single channel, with any other functions (mixing,
34
quench, etc.) to be performed up- or downstream of the device. For ease of handling, the
reactor was to be rectangular, and as wafer handling during fabrication proscribes
placement of features closer than 1 cm to wafer edge, the design was to be a square
inscribed in a 13-cm-diameter circle.
The channel depth was selected to be 725 μm, thus resulting in channel width of 1.5
mm, as discussed for the microreactor design. This allowed 57 channels of 9-cm length
to be packed next to each other, with 100-μm separation, resulting in a 5-mL reactor, an
order of magnitude larger than the largest Goldilocks device. Figure 2.10 compares the
intended reactor layout and an actual device.
Fabrication method and techniques
The detailed process run sheet for the KOH etching process is given in Appendix A.
In brief, the procedure for the fabrication consisted of mask layer deposition, lithography,
etching, protective layer growth or deposition, and bonding.
To serve as a mask layer, 200 nm of LP-CVD silicon nitride was deposited on the
surface of virgin silicon-(100) wafers (Silicon Quest International, Inc., Santa Clara, CA)
with flats cut along the <110> direction. Photolithography was performed using 1 μm of
thin photoresist (OCG 825 positive resist), following the standard operating procedure
(SOP) using an Electronic Visions EV620 Mask Aligner. It is critical during the UV
exposure to align the photomask to the crystal plane of the wafer. As can be seen in
Figure 2.9, the mask has a number of closely spaced horizontal slits at the region of the
wafer flat. By looking through those slits on the mask, it is possible to view and align the
wafer.
For wafers containing features on both sides, the two lithography steps were
performed sequentially prior to nitride etching. To do so, photoresist coated on one side
of the wafer was pre-baked (Blue M Model DDC-146C oven, 95ºC) for only half of the
time prescribed by the SOP (15 minutes instead of 30), followed by coating of the other
side and a full pre-bake time. It is best to first coat the side with more critical features
(i.e., channels), as opposed to the side with features such as ports.
After exposure of the front side, the wafers were developed for 20% of the SOPprescribed development time (12 seconds out of 60, in OCG 934 1:1 developer),
35
sufficient to make visible the features, including alignment marks. After exposure of the
second side, development was performed until all features appeared fully developed
when viewed under the fluoroscope. Following development but prior to post-baking, the
edges of the wafers were “painted” with photoresist using swabs to protect the nitride at
the rim. This prevents the rim of the wafer from being etched in KOH, which would
result in a jagged edge, making the wafers more brittle and difficult to handle. Optimally,
wafers would be developed as a batch, using a Teflon cassette to submerge them.
However, if it was desired to develop one wafer at a time, care was made to ensure flow
of developer to both sides of the wafer (i.e., the wafer was not permitted to lie flat on the
bottom of the developer dish).
When performing two-sided lithography, great care must be taken not to inadvertently
scratch or damage the resist on either side, as this would result in flaws during etching.
Alternatively, it is possible to perform lithography and nitride etching on one wafer side
at a time. This requires more processing time, although it is easier to handle wafers that
only have process-critical resist on one side.
The nitride mask layer was etched using RIE (Lam Research Model 490B Reactive
Ion Etch), following an SOP. After removal of the photoresist (piranha solution, 10
minutes), the wafers were placed into a 25 wt% KOH solution (aqueous) at 80ºC. This
resulted in etch rates of approx. 60 μm/hr, varying slightly based on the silicon doping,
resistivity, and possible deviations on bath temperature. After 1 hr of etch time, the
wafers were removed, and features were measured to more precisely determine the etch
rate and calculate the etch time. Measurement was done under an optical microscope,
comparing the depths of the top and bottom of a vertical side wall, doing so at several
places on the wafer. One hour prior to calculated etch end, this was repeated, and the
final etch time was determined. When wafers from multiple processes were etched
simultaneously, the final-hour etch measurement was performed for each process’s
wafers, and as the less deeply etched wafers were completed, they were removed,
allowing other ones to be etched further. In this fashion, several processes’ wafers could
be etched at the cost of a single etch step.
To ensure uniform etching with smooth channel side walls and bottoms, wafers were
oriented vertically within the bath. This allowed the large amounts of evolved hydrogen
36
gas to easily escape. Otherwise, if the wafers were placed horizontally, H2 was able to
become trapped between wafers or inside features, causing non-uniform etching across
the wafer, as well as local depletion of KOH between gas bubbles and silicon, resulting in
rough surfaces due to microscopic pyramidal protrusions and pits. When the wafers were
placed vertically, it was very important to ensure that the water level of the heating bath
is at least 2-3 cm above the liquid level inside the KOH tank, providing more uniform
temperature within the KOH solution. Additionally, a layer of air-filled plastic spheres
was floated atop the heating bath water to afford thermal insulation. The KOH bath was
covered to prevent water evaporation and changes in concentration.
After properly cleaning the wafers, the nitride layer was removed by submersion in
phosphoric acid at 165ºC. To afford chemical compatibility and resistivity, 500 nm of
wet thermal silicon oxide was grown on the wafers (MRL Industries Model 718 System
Atmospheric Oxidation Furnace, 90-minute wet oxidation).
Alternatively, for more
robust protection, 500 nm of LP-CVD silicon nitride was deposited (Thermco 10K 4Furnace System). Subsequently, the wafers were bonded to borosilicate glass wafers
(Silicon Quest International) via anodic bonding at 400ºC and 800 V (Electronic Visions
EV620-501 Wafer Aligner/Bonder), with voltage maintained until current across the
wafers decreased to below 0.5 A. For wafers with no port holes etched, a glass wafer
with holes bored through with an excimer laser was used (Resonetics Excimer Laser
Drill).
Microreactor packaging
Bonding pads (deposited by a Temescal Semiconductor Products Electron Beam
Evaporator) were incorporated in the design to allow for solder-based packaging that we
have previously developed.31 For this reason, the ports were placed in the same locations
as in the DRIE-etched devices for which we have developed tube alignment components
that also served as fluidic interfaces to those tubes. This allows the use of already
existing components for ease of application.
While solder packaging has a great deal of utility, metal deposition is a high-cost
fabrication step. Thus, to allow the elimination of said step from the process and to
permit a fully reversible packaging process, a compression-based fluidic interface
37
(“chuck”) was made by the MIT Central Machine Shop.
To retain the Goldilocks
flexibility and taking advantage of the identical port layouts, a single chuck was made to
accommodate all three reactor sizes (Figure 2.11a,b). Ports were drilled to be ¼-28
interfaces for standard fittings (flat-bottom nut-and-ferrule combination, P-200 and P201, Upchurch Scientific, Oak Harbor, WA). To allow it to function as a heater, holes
were drilled in its bulk for cartridge heaters. Stainless steel was used for the chuck to
provide better chemical compatibility.
To equally distribute the stress from the compressing top, inserts were made to fill the
remaining space when using the medium and small reactors (Figure 2.11c).
When
compressing the large reactor, the torque at the o-rings would be sufficient to crack the
device. For that reason, indentations for sets identical o-rings (Viton® size 004, part
1201T14, McMaster-Carr, Aurora, OH) were placed at each of the four quadrants of the
chuck, distributing the stress on the largest device.
(a)
(c)
(b)
Figure 2.11. Goldilocks compression chuck: chip (a) and fitting sides (b); metal resizing insert (c).
Mesoscale reactor packaging
The large area of the reactor allows for a wide choice of packaging methods. For
applications at temperatures not exceeding 125ºC, Nanoports (Upchurch® N-333) are
simple to apply and highly useful.
However, for higher temperatures, compression
packaging can be applied, similarly to a previously developed design.51 A symmetrical
compression chuck was developed to allow a single design to be applied to either side of
the reactor (Figure 2.12a). Holes were bored through the chuck to allow for coolant flow
and permit high temperatures throughout most of the chip while maintaining tolerable
temperatures at the polymer o-rings and fittings. For heating, a simple aluminum plate
38
was designed to fit between the two compression chucks, accommodating two 100-W
cartridge heaters (Omega® CSS-034100/120V). The reactor was to be held down against
the heating plate by a piece of glass via two aluminum hinge pieces. Figure 2.12b shows
a fully assembled device with the heating plate and compression pieces. Section 2.3.2
discusses the results of application of the KOH-etched meso-reactor.
(b)
(a)
Figure 2.12. Meso-reactor compression chuck (a) and a fully assembled meso-reactor (b).
2.2.2 HNA-Etched Reactors
Mesoscale reactor design
To produce highly robust mesoscale fluidic devices more rapidly than is possible with
KOH etching, the HNA wet etch was applied, as it is capable of producing 600-μm-deep
features within 10 minutes of etch time and etches all crystal planes at equal rates. The
latter feature allows for a highly flexible design of layouts, as it is not limiting to etching
along crystal planes as caustic etches would be. Additionally, it produces features that
are nearly semicircular in cross-section, which greatly reduces stress concentration,
producing a more pressure-robust device than the KOH-etched one and reducing axial
dispersion as compared to square channels (see sections 2.3.2 and 2.3.3 for discussion).
Similarly to the KOH-etching technique, the HNA method was applied to 1000-μmthick wafers to maximize the possible reactor volume. Once again, a single channel was
produced, intending all other functions to be performed outside of the device. As the
39
feature layout is flexible, a nested spiral design was made, spiraling a channel inward
from the rim of the wafer to its center, followed by an inversion of the spiral to lead back
from the center to the rim. This most closely mimics the familiar to most users coiled
tube, avoiding any sharp turns or bends and reducing the likelihood of clogs due to
precipitates. Additionally, such a double spiral allows the placement of the inlet and the
outlet in close proximity for ease of packaging.
Considering the 1 cm rim to the wafer edge for processing, the design was a spiral
fitting within a 13-cm-diameter circle. To produce a 5-mL device, similar to the KOHetched meso-reactor, the channel depth was selected to be 600 μm, thus resulting in
channel width of 1.25 mm at the top, with a nearly semicircular cross-section (the feature
width was 50 μm, as with the KOH designs). Figure 2.13 shows the designed layout and
an actual device, packaged with Nanoports.
(a)
(b)
Figure 2.13. Spiral mesoscale silicon reactor layout (a) and actual device packaged with Upchurch
Nanoports (b).
Fabrication method and techniques
The detailed process run sheet for the HNA etching process is given in Appendix A.
In brief, the procedure for the fabrication was nearly identical to that of KOH etching and
consisted of mask layer deposition, lithography, etching, protective layer growth or
deposition, and bonding, using the same equipment.
Different mask layer thicknesses were explored (see section 2.3.2 for the results). A
layer of 2 μm of LP-CVD silicon nitride was used for the deepest etches. Because HNA
40
is not crystal-plane-specific, the orientation of the wafer flat was immaterial for this
process, as was alignment of the mask to the flat. Photolithography was performed in an
identical fashion to that for KOH-etched wafers, with a few minor differences. Thick
photoresist (AZ 9260) was used, with 10-μm thickness, and following its respective SOP
for coating, exposure, and development (with AZ 440 developer). Two-sided lithography
is also possible here if through-holes are desired.
After lithography, the nitride mask layer was etched using a 5-step recipe of RIE
interspersed with cooling, following a previously developed procedure.
Without
removing the photoresist, the wafers were placed into an HNA solution equipped with a
magnetic stirrer atop a stirplate at room temperature. For study purposes, the HNA
composition was varied (see section 2.3.2 for the results). The optimal composition was
found to be 6:3:1 volumetric ratio of HF (49%) / HNO3 / C2H5COOH (glacial). This
resulted in an etch rate of 60-100 μm/min, varying based on temperature and agitation.
Etch rate was measured after 1 min and after 3 min, as well as at intervals of 30 seconds
after 5 min, with wafers being rotated relative to the cassette after each measurement.
After each removal, the wafers were dipped into clean water. Large amounts of reddishbrown fumes (toxic NOx gas) were evolved during the etching process. Thus, for deep
etches, it is crucial to have appropriate safety precautions and ample ventilation.
Similarly to the KOH process, the most uniform etching was achieved when wafers
were oriented vertically within the bath. Even so, at these etch rates, the etch-formed
surface was roughened. To eliminate the roughness, following the completion of the
etch, the HNA solution was diluted by a factor of 2 with acetic acid, and the wafers were
submerged for an additional 30 seconds. Thus, the etched channels were well polished.
Removal of nitride, protective layer formation, and bonding to glass were identical to
corresponding steps for KOH etching. For wafers with no port holes etched, excimerlaser-drilled glass wafers and unprocessed glass wafers were used. With unprocessed
glass wafers, holes were later made using a Dremel drill (300 series, Dremel®) on a
vertical mount (model 220, Dremel®), equipped with 0.8-mm diamond-coated mill bit
(W754730, Wolfco Inc., Pomfret, CT).
41
Mesoscale reactor packaging
Similarly to the KOH-etched meso-reactor, a wide choice of packaging methods
exists for the HNA-etched device. Nanoports are highly versatile for many desired
applications and are used in most cases. However, to achieve temperatures above 120ºC,
compression packaging was designed. A symmetrical chuck was developed to allow a
single design to be applied regardless of whether the through-holes of the device were
made in the silicon or the glass layer. This chuck also contained holes for coolant flow to
permit high temperatures throughout most of the chip while maintaining tolerable
temperatures at the polymer o-rings and fittings. Because compression was applied only
at one end of the chip, heating could be applied much more easily by either resting the
free part of the device on a hotplate, silicon side down, or submerging it in an oil bath.
2.3
Results
2.3.1 KOH-Etched Microreactors
The KOH etching process to produce vertical-side wall features and features of
different depths within the same etch time was successfully demonstrated. Within the
same etchant tank, up to 15 wafers were processed simultaneously, most of them having
features on both sides, thus verifying the utility of the KOH etching method to efficiently
and rapidly fabricate silicon devices.
Successful etching of KOH-etched microreactors was achieved, with reactors on the
same wafer having features of 76, 95, 114, and 400 μm deep, as well as through-holes
(etched in 650-μm-thick wafer). Figure 2.14shows the three Goldilocks devices side-byside, along with their dimensions. As discussed in section 2.2.1, the <110>-aligned
features with the largest corner compensation features experienced the least corner
overetch. Figure 2.15 shows the mask features and the etched corners for the Goldilocks
reactor set. The <100>-aligned features, though, were formed as expected, with sharp
90º convex corners and with concave corners “banked” be 54.74º sloping side walls
positioned 45º to the channel.
42
Figure 2.14. Goldilocks reactor set.
(a)
(b)
(c)
Figure 2.15. Etched convex <110>-aligned corners after equal etch times: (a) 200-μm channels; (b)
250-μm channels; (c) 300-μm channels.
When wafers were positioned vertically within the bath, the etching was quite
uniform, with a maximum etch depth difference of <2% across the wafer and <1%
between wafers when measuring at the same position, with total etch depths of up to 750
μm. With etching in this orientation, etched side walls were found to be very smooth,
with no roughness at scales down to below 1 μm, as seen by scanning electron
microscopy (SEM, Zeiss® Supra 40). SEM images of a channel bottom show particles
that may be dust accumulated due to exposure to the environment and do not appear to be
features of the surface (Figure 2.16).
43
Figure 2.16. SEM image of KOH-etched reactor bottom.
(a)
(b)
Figure 2.17. Reactor flaws resulting from (a) feature underdevelopment and (b) scratched resist.
The etch process is highly sensitive to flaws in the lithography. Small flaws produced
by occasional careless handling (scratch in the resist) or imperfectly developed features
over very small areas become greatly amplified by combinations of etching and etch-stop
planes. As shown in Figure 2.17a, a micron-scale underdevelopment leads to a channel
etch being terminated and bounded in the middle, resulting in a dead-ended channel.
This can be mollified be excimer-laser ablation, at additional cost and processing time, to
44
breach the formed dam to an extent. Conversely, a slight widening of a feature or a
scratch between features, due to the lateral etching, will extend horizontally to breach a
wall between two channels (Figure 2.17b), causing significant channeling or bypassed
channels.
All of these flaws must be avoided via meticulous wafer handling and
monitoring of photoresist development.
The microreactors have been successfully compression-packaged and were shown to
be robust at pressures of 7 bar. While having good performance at homogenous liquid
flow, the wide turns proved to be a hindrance to gas-liquid flow, allowing small gas
bubbles to become trapped at the wide corners. However, many different designs, with
less sharp turns (i.e., wider channel walls) and narrower turns, can be produced
depending on the desired application.
2.3.2 KOH-Etched Mesoscale Reactors
Using the same process as that for the KOH-etched microreactors, mesoscale reactors
were fabricated. Identical constraints and requirements for careful handling apply to the
mesoscale design. The meso-reactors were characterized regarding their flow profile and
dispersion, as well as thermal profile when packaged as previously shown in Figure
2.12b. Temperature was measured by an IR gun (Optris® LaserSight) calibrated against
a 204ºC lacquer (Omega® Omegalaq 400 F).
To transfer knowledge obtained on the microscale, it is necessary to have a full
understanding of scaling phenomena. As the first step towards that goal, dispersion was
calculated by modeling the reactor as a long rectangular channel. At laminar flow
regimes (which would be the case for all conceivable liquid-based applications in these
devices), the axial dispersion coefficient D is given as:52
u 2d 2
D =D +
192D
2.3
where D is the diffusion coefficient, u is the linear velocity, and d is the equivalent
diameter (for a rectangle of 1.55 × 0.75 mm, d = 1.05 mm). D for acetone in water found
in literature53 was used. At flow rates of 0.5 and 1.0 mL/min, D is calculated to be
2.95×10-4 and 1.18×10-3 m2/s, respectively. The axial dispersion Péclet number, given as:
45
Pe Ax =
uL
D
2.4
with L being the length of the tube, 4.3 m in this case. PeAx for the two aforementioned
flow rates is 104 and 53, respectively; thus, at 0.5 mL/min, the plug flow approximation
can still be used, but at higher flow rates, axial dispersion becomes significant.
Next, dispersion was experimentally evaluated in the meso-reactors via a residence
time distribution (RTD) of a step injection (via a 6-way valve, Upchurch® V-451) of a
tracer (0.2 vol.% acetone in water, detected by UV-Vis spectroscopy in a Waters® 2996
Photodiode Array Detector at 280 nm, using EmpowerTM software). For the valve/tubing
setup without the inclusion of the reactor, the resulting concentrations and first moments
as functions of time are shown in Figure 2.18a and Figure 2.19a for 0.5 and 1.0 mL/min
flow, respectively. The RTD and first moments for the reactor (with the valve setup) are
shown in Figure 2.18b and Figure 2.19b for 0.5 and 1.0 mL/min flow, respectively.
Additionally, the RTD of the valve setup was convolved with the theoretical first moment
of the reactor, calculated based on the dispersion coefficients D calculated above, using
the following relation, valid for open-open vessels with axial dispersion:52
u
Et =
4π Dt
e
−
( L −ut )2
4 Dt
t
Cout (t ) = ∫ Cin (t ')E(t − t ')dt '
0
2.5
2.6
where t is time and Cin is the concentration leaving the valve setup. For the flow rates of
0.5 and 1.0 mL/min, Figure 2.18c and Figure 2.19c, respectively, show thus calculated
RTDs and their first moments, demonstrating very good agreement with the experimental
results. Therefore, the axial dispersion model is valid for this device.
As this experiment demonstrates, dispersion is fairly significant here, as is expected
for a rectangular channel of this length. The average volume of the reactor, as measured
by mean residence time (subtracting that of the system itself), is just under 5 mL. The
relative standard deviation (calculated as the ratio of square root of the variance to the
mean residence time), subtracting the variance due to the setup, was 11.1% and 12.9% at
0.5 and 1.0 mL/min flow, respectively. At the investigated flow rates, a change in
reaction conditions requires a wait of at least 1.6 reactor residence times prior to
achieving steady state, with 0.50-0.70 residence times required to account for dispersion.
46
5×10-2
45
-2
4×10
35
30
-2
3×10
25
20
C(t)
15
E(t)
-2
2×10
10
E(t)
Adsorbency * 100
40
-2
1×10
(a)
5
0
0
50
0
150
100
Time (s)
45
-3
40
7×10
45
(b)
6×10
35
C(t)
30
E(t)
-3
5×10
-3
4×10
20
3×10
E(t)
25
-3
15
-3
2×10
10
-3
1×10
5
0
200
400
600
800
(c)
-3
6×10
35
C(t)
30
E(t)
-3
5×10
-3
25
4×10
20
3×10
-3
15
-3
2×10
10
-3
1×10
5
0
0
-3
7×10
0
0
1000
E(t)
Adsorbency * 100
40
Adsorbency * 100
-3
0
200
400
Time (s)
600
800
1000
Time (s)
Figure 2.18. Exit concentrations with time C(t) and first moments E(t) of the valve setup (a), and the
KOH-etched meso-scale reactor experimental (b) and calculated values (c) at 0.5 mL/min.
-2
8×10
40
7×10
35
6×10
-2
-2
30
-2
5×10
25
20
C(t)
15
E(t)
4×10-2
E(t)
Adsorbency * 100
45
-2
3×10
-2
2×10
10
-2
5
(a)
0
0
20
40
60
80
1×10
0
100
Time (s)
-3
45
40
-3
45
C(t)
25
-3
3×10
-3
2×10
15
10
-3
1×10
5
-3
4×10
35
30
C(t)
25
-3
3×10
E(t)
20
-3
2×10
E(t)
E(t)
20
Adsorbency * 100
30
5×10
(c)
40
-3
4×10
35
E(t)
Adsorbency * 100
5×10
(b)
15
10
-3
1×10
5
0
0
0
0
200
400
600
0
0
Time (s)
200
400
600
Time (s)
Figure 2.19. Exit concentrations with time C(t) and first moments E(t) of the valve setup (a), and the
KOH-etched meso-scale reactor experimental (b) and calculated values (c) at 1.0 mL/min.
47
As discussed in section 2.2.1, a modular heater was machined for the meso-reactor,
comprised of an aluminum base and a glass top, with a graphite heat-spreading layer
inserted between the chip and the aluminum plate. The two holes drilled through the Al
plate accommodated two 100-W cartridge heaters (Omega® CSS-034100/120V). A
small hole was drilled in the middle of the plate, designed to position a thermocouple
under the center of the reactor, 0.5 mm beneath the surface of the Al plate.
The
compression chucks were drilled through for coolant flow to prevent overheating of
polymer fittings and o-rings. The system was tested using a PID feedback controller to
operate the heaters based on data from the thermocouple. To produce a temperature of
200oC, the setpoint was chosen as 210oC to account for a thermal gradient. Coolant
water at 20oC was flowed through both of the compression pieces. Figure 2.20 shows the
temperature values at different locations on the reactor. It can be seen that near the
compression pieces, the temperature is much lower than the setpoint, which is to be
expected. Approximately 10% of the reactor volume will be at a temperature more than
10oC below the setpoint at these conditions. However, this can be attenuated by using a
higher-temperature coolant flow, reducing the thermal gradient.
Figure 2.20. Thermally packaged meso-scale reactor, with temperature values (ºC) when heated to a
setpoint of 210ºC, with compression chucks cooled to 20ºC.
The KOH-etched meso-reactors have been found to be highly fragile, forming cracks
with flows at any significant pressure drop. The cracks always formed along a channel
48
wall, running lengthwise along the reactor.
This is believed to be due to stress
accumulation along a crystal plane, concentrated at a pinch point formed by the corner of
a channel wall and floor. Attempts were made to reinforce the silicon side of the mesoreactor by anodically bonding it to another borosilicate glass wafer.
While this
successfully reinforced the silicon, when flow was applied to the reactor at somewhat
elevated pressures, cracks invariably formed in the top glass wafer. These cracks, once
again, were nearly perfectly linear and always occurred along an edge of a channel wall.
It is believed that the difference in coefficients of thermal expansion between the
silicon and glass is the primary cause of this fragility.
-6
-6
The coefficient of thermal
-1
expansion of silicon is varies from 3.8×10 to 2.6×10 ºC between 400ºC and room
temperature,54 while that of Borofloat® 33, the glass used for anodic bonding, has this
coefficient as 3.25×10-6 ºC-1 between room temperature and 300ºC. This glass is used
because its thermal expansion is most similar to that of silicon at bonding temperatures.
However, there remains a compression of the glass by approximately 3×10-4 of its total
length in any direction.
The stress in the glass, which varies linearly with strain,
proportionally to Young’s modulus E (for Borofloat® 33, E = 64 GPa), thus becomes 20
MPa, which is not an insignificant value but one that is nonetheless far lower than 69
MPa, the flexural strength of this borosilicate glass.
For microreactors, where the dimensions are fairly small and the etched area of the
silicon is not nearly as large relative to the fully supported rim, this stress is distributed
well to the bonded areas and is thus well within the capability of the glass. On the other
hand, for the meso-reactor, the strain is large relative to the fully supported reactor edge;
in addition, a very large area of the glass is unsupported by silicon, and the supports are
very narrow and straight. This combines to them acting as fulcrums, against which, with
any application of force normal to the glass (i.e., pressure from liquid in channels), the
glass can release its strain by cracking. To resolve this problem, it is necessary to have
wider channel walls to bond to the glass, as well as to alter the layout so as to avoid long
features in any one direction. As KOH etching is particularly apt at forming long
rectangular features, it is not ideal for such a purpose. Therefore, the flexibility afforded
by HNA etching is preferred for meso-scale silicon-glass reactors.
49
2.3.3 HNA-Etched Mesoscale Reactors
Initially, tests were made to determine the proper mask thickness and HNA
composition to allow for a sufficiently deep etch to produce meso-reactors. It was found
that at faster etch rates, the selectivity of the etchant for silicon over nitride improved. At
the most frequently reported composition of 5:10:16 by volume of HF / HNO3 /
C2H5COOH,48 the etch rates of silicon and nitride were 20 μm/min and 60 nm/min,
respectively, within reported ranges. While 2 μm of nitride should theoretically be
sufficient for a desired 600 μm etch, a mask should be sufficiently thick to provide
protection against a certain amount of overetch or etching variability. Thus, a faster etch
was desired.
According to the literature, the fastest etch rate of 250 μm/min is produced with 2:1
HF/HNO3 with no dilution.55 This was confirmed experimentally, and the silicon nitride
etch rate was approx. 500 nm/min on the front side. However, when using this etch, the
nitride on the back side of the silicon wafer (2 μm in thickness) was completely etched
away after 2.5 minutes in the same pattern as the silicon features on the front side,
although no such pattern was deposited on the back side. This is caused by the large
exothermicity of the etching reaction: within the silicon channels, the reaction occurs at
increasing proximity to the back side, with the silicon very effectively conducting the
heat. If the solution does not dissipate the thermal energy sufficiently rapidly, the nitride
on the back side below the etched pattern experiences increased temperature and
accelerated etch rates. Thus, the composition providing the maximum etch was not
usable for meso-reactor fabrication.
By diluting the 2:1 HF/HNO3 solution to 90% strength with acetic acid, a controllable
etch rate was produced. The silicon and nitride etch rates were approximately 100
μm/min and 250 nm/min, respectively. This allowed for a process that would produce
600-μm-deep features without etching through the mask layer if 2 μm of nitride are used.
Crucially, the isotropic etch has identical constraints and requirements for careful
handling during photolithography and mask etching as those for KOH fabrication.
Nested-spiral mesoscale reactors were successfully fabricated using the HNA etching
method following the procedure discussed in section 2.2.2. As expected, etched features
were nearly semicircular, with lateral etching being equal to vertical etching at all points
50
on the reactor. The optimal etch uniformity across the wafer was found to occur when
only one wafer was etched at any given time, using about a 7-cm depth of HNA solution
and positioning the wafer horizontally such that it is in the middle of the solution, with
the primary features to be etched (the channels) facing upwards. While very large
amounts of NOx gas were formed, the bubbles were easily released from the features.
Nonetheless, the etch rates varied by approx. 10% from wafer edge to center, resulting
feature cross-sectional areas varying by as much as 20% across the wafer.
The meso-reactor has been demonstrated to be robust to pressures of 6.8 bar (100-psi
backpressure); however, at pressures of 17 bar (250-psi backpressure), the glass layer
experienced failure, even when thicker wafers were used. Thus, it is safest to apply the
silicon meso-reactor at pressures of or below 6.8 bar. To achieve greater pressure
robustness, based on the flexural strength of the glass, this limits the ratio of wall area to
etched area to 1:1, which in turn limits the meso-reactor to a volume of 2.4 mL.
At the applied etch solution composition, the etched features were found to have a
rather rough surface, with roughness on the order of several microns. This is due to the
large amounts of gas that was formed, leading to a great deal of local variation of HNA
concentration. As the bubbles formed, they limited mass transfer around them, leading to
microscopic irregularities. However, this was significantly ameliorated by submerging
the etched wafers for 30 seconds into the same HNA diluted 1:1 with acetic acid. As a
result, the surfaces became much smoother, with less roughness than is observed on
DRIE-etched side walls.
Similarly to the KOH-etched mesoscale reactors, dispersion calculations were performed
for the HNA ones, as well as experimental RTD analyses. Based on equations 2.3 and
2.4, using a diffusivity value of toluene in methanol found in literature56 and the
equivalent diameter for a semi-cylinder with 0.6-mm radius as d = 0.9 mm, at flow rates
of 0.5 and 1.0 mL/min, the axial dispersion coefficient D was calculated to be 9.16×10-4
and 3.66×10-3 m2/s, respectively. With the channel length of 8.3 m, the PeAx values for
the two aforementioned flow rates are 133 and 67, respectively. Thus, for a reactor of a
similar volume, the HNA-etched spiral device results in less dispersion than does the
KOH-etched device. However, at flow rates measured in mL/min, dispersion still plays a
significant role, as would be expected for an 8.3-m-long channel.
51
For these reactors, a 5-μm pulse injection of a tracer (10 vol.% toluene in hexane,
detected by UV-Vis spectroscopy) was used, delivered via a sample loop of a 6-way
valve. For the valve/tubing setup without the inclusion of the reactor, the resulting
concentrations as functions of time are shown in Figure 2.21a and Figure 2.22a for 0.5
and 1.0 mL/min flow, respectively. The RTD and first moments for the reactor (with the
valve setup) are shown in Figure 2.21c and Figure 2.22b for 0.5 and 1.0 mL/min flow,
respectively. Once again, the RTD of the valve setup was convolved with the theoretical
first moment of the reactor, determined using equations 2.5 and 2.6 based on the
calculated dispersion coefficients D. These are shown in Figure 2.21c and Figure 2.22c
for the flow rates of 0.5 and 1.0 mL/min, respectively. Different integration methods
were used between the valve RTD experiments and the those including the reactor,
resulting in a different maximum peak height for the convolution. However, the overall
trend demonstrates good agreement with the experimental results.
60
Adsorbency
50
40
30
20
10
(a)
0
0
20
40
60
80
Time (s)
5×10-3
(b)
-3
1.5
-3
1×10
0.5
0
0.0
5×10 -3
0.6
4×10 -3
0.5
3×10 -3
C(t)
E(t)
0.4
E(t)
1.0
E(t)
2×10
C(t)
E(t)
6×10 -3
0.7
3×10-3
-3
7×10 -3
(c)
0.8
4×10
2.0
Adsorbency
0.9
Adsorbency
2.5
2×10 -3
0.3
0.2
1×10 -3
0.1
0
0
0
200
400
600
800
1000
0
Time (s)
200
400
600
800
1000
Time (s)
Figure 2.21. Exit concentrations with time C(t) and first moments E(t) of the valve setup (a), and the
HNA-etched meso-scale reactor experimental (b) and calculated values (c) at 0.5 mL/min.
52
20
18
16
Adsorbency
14
12
10
8
6
4
(a)
2
0
10
2.5
(b)
0.35
8×10 -3
0.30
1.0
2×10 -3
0.5
40
1×10-2
(c)
8×10-3
6×10-3
C(t)
E(t)
0.20
4×10-3
0.15
2×10-3
0.10
0
0
0.05
0.0
E(t)
4×10 -3
30
0.25
6×10 -3
C(t)
E(t)
1.5
1×10 -2
E(t)
Adsorbency
2.0
20
Time (s)
Adsorbency
0
0.00
0
100
200
300
Time (s)
400
0
500
100
200
300
Time (s)
400
500
Figure 2.22. Exit concentrations with time C(t) and first moments E(t) of the valve setup (a), and the
HNA-etched meso-scale reactor experimental (b) and calculated values (c) at 1.0 mL/min.
The reaction volume, based on mean residence time, was 5 mL. The relative standard
deviation (calculated as for the KOH meso-reactor, section 2.3.2) was 11.0% and 14.2%
at 0.500 and 1.0 mL/min of flow. At the investigated flow rates, a change in reaction
conditions requires a wait of at least 1.6 reactor residence times prior to achieving steady
state, with 0.50-0.70 residence times required to account for dispersion.
2.4
Conclusions
Wet-etch microfabrication has been demonstrated to be a highly versatile alternative
to expensive and time-consuming plasma etch techniques. These methodologies allowed
the simultaneous two-sided processing of up to 15 wafers, as well as very rapid etching to
large depths. Devices produced with these methods have been shown to be robust to high
temperatures and pressures, making them applicable to a wide range of continuous-flow
chemical studies. Importantly, this fabrication methodology has proven to be rapid and
simple to execute at much lower processing costs, providing an inexpensive means of
quickly producing a large number of silicon devices.
53
Chapter 3.
Interdigitated Micromixers†
Many chemical synthesis steps are extremely rapid and highly concentrationdependent. To be able to study the kinetics of such reactions, it is very important to be
able to rapidly mix the starting reagents, as well as to quickly terminate the reaction by
addition of a chemical agent. This can be done best in using continuous flow and
incorporating inline mixers to rapidly eliminate concentration gradients as two or more
streams are merged into one.
A modular silicon micromixer based on the principle of flow interdigitation and
focusing was designed and fabricated for high-flow rapid mixing at a wide range of
reaction conditions. The goal was to design an efficient micromixer that is low in
volume, permitting fast kinetic analysis, and with a reasonably low pressure drop,
allowing pumping at high flow rates for maximal production through each system. In
addition, the mixer design had to be sufficiently simple and cheap to manufacture in
order to be practical for industrial applications. To best satisfy the above conditions, an
interdigitated laminar micromixer was designed.57, 58
The mixer operates by splitting two inlet flows into a large number of channels,
interdigitating them, and constricting the laminated flow to create sub-micron diffusion
lengths.
A second-generation redesign of the micromixers was performed, greatly
simplifying the fabrication procedure, decreasing the pressure drop across the device by a
large factor, and introducing a design to mix three streams into one, laminated in an
ordered fashion. For each of the fabricated micromixers, mixing was quantified using the
Villermaux/Dushman method of competing reactions, with UV-Vis detection of the
photoactive species, and compared against a commercial micromixer.
†
This chapter describes work done in close collaboration with Edward R. Murphy and
Jason G. Kralj, who at the time were fellow doctoral students in the laboratory of Prof.
Klavs F. Jensen.
54
3.1
Continuous Micromixing
Mixing is a highly important phenomenon for continuous-flow chemistry studies, as it
is often necessary to combine reagents for a chemical synthesis to occur, as well as to
terminate a reaction by addition of another chemical species. For studies of chemical
reaction kinetics, it is important that the residence time that is measured represent the
time of reaction itself, when the reaction stream is fully mixed. Thus, it is necessary that
the mixing time of incoming be several orders of magnitude less than the reaction time
for accurate kinetic study. Similarly, if the reaction termination is to occur by dilution or
by introduction of a quenching chemical agent, the mixing of the quench stream with the
reaction medium must be equally rapid. Thus, being able to rapidly mix reagents in a
continuous fashion for microscale flow chemistry is very significant.
Most commonly, mixing has been defined as minimization of the intensity of
segregation, particularly for binary mixtures of liquids.59 On the microscale, liquid flows
are nearly always laminar, with no turbulence.
Thus, mixing is effected solely by
molecular interdiffusion.60 To improve mixing, it is therefore necessary to either increase
the area of contact between the mixing streams or to decrease the effective diffusion
length (or both). The dominance of interfacial properties on the microscale allows for a
wide range of micromixer designs in a variety of materials, including metals, polymers,
glass, and silicon, the latter two being the most prevalent for micromixer fabrication.
Such designs operate by either active mixing (via the external input of energy) or passive
mixing (restructuring the flow to maximize interaction). Comprehensive reviews and
discussions of various micromixer designs, both active (ultrasonication, thermal
disturbance, microimpellers, etc.) and passive (lamination, injection, split-recombine,
etc.) are available in literature.61,62
Many microfluidic devices designed for chemistry studies incorporate a mixing zone
as part of their design. These mixing zones frequently arrange the incoming flows into
the shapes of a T-mixer63 or a Y-mixer,64 or shape the flow channel to augment mixing,65
with some designs additionally providing flow splitting and lamination to improve
mixing.29,
66
Mixing occurs rapidly in the narrow channels that typically follow the
junctures. However, these mixing zones may not be sufficient for many reactions with
55
rapid kinetics. In those cases, a modular micromixer device may be necessary to provide
enhanced mixing.
To reduce the required footprint and auxiliary equipment, a passive micromixing
design was selected, as it only requires the micromixer itself to operate, thus being more
practical and relatively easier to manufacture and operate. An interdigitated laminar
mixing principle with flow focusing was used due to its ability mix rapidly and efficiently
with a relatively low pressure drop.67,68 By splitting the flow into a large number of
laminae and compressing the laminar stack into a narrow channel, the diffusion length is
radically reduced. Additionally, the availability of many laminae for each component
flow ensures that this is accomplished with minimal pressure drop.
3.2
Micromixer Design
3.2.1 Three-Wafer Stack
The initial micromixer design was intended to rapidly mix two incoming streams of
liquid by splitting the streams, interdigitating them, and focusing the combined stream
into a narrow channel, where the bulk of the mixing occurs. Figure 3.1 shows the
illustrations and photograph of the mixer, showing the three wafers: the primary
processed silicon wafer sandwiched between a drilled and a partial borosilicate glass
wafers. The two primary design conditions were the rate of mixing and the ability to
evenly distribute the main flow across the manifold that splits the streams. As previously
discussed, for laminar flow, the rate of mixing is determined by the diffusion time, given
in Chapter 2, equation 2.1. Thus, splitting incoming streams into as many lamina as
possible and focusing them as narrowly as possible will maximize mixing. The flow
distribution is accomplished by having the pressure drop along the manifold be negligible
compared to the pressure drop following the manifold. A difference of two orders of
magnitude was considered to satisfy this criterion.
For compactness, the mixer footprint was selected to be 25 mm × 20 mm. To provide
rapid mixing, it was decided to split the two incoming flow streams into 50 and 51
lamina, respectively. The focusing was achieved in a triangular cavity, which narrows to
a width of 500 μm, compressing the formed laminae to slightly below 5 μm each and
56
creating a diffusion length of 2.5 μm. According to work by Schönfeld and co-workers,
this design would ensure mixing on the order of 10 ms for a diffusivity of 10-9 m2 s-1, a
representative value for ions and other diffusive species in aqueous media.68, 69
(a) (c) (b) (d) Figure 3.1. Interdigitated silicon micromixer – (a) three dimensional rendering with (b) a close-up of
corner of distribution channels showing the two flow streams being split into 50 and 51 interdigitated
channels, (c) a close-up of the end of the interdigitated channel section forming 101 lamiae that are
subsequently compressed in the converging channel; (d) photograph of actual device.
The focuser cavity and channel, flow distribution manifolds, and through-holes were
etched using DRIE. Rectangular post features were added to the focusing cavity to
reduce the apparent feature size for DRIE, ensuring a uniformly deep etch across the
cavity. Additional benefits are the reduction of mixer volume and the possibility for
improved mixing due to flow splitting and recombination around the posts. The initial
channels through which the fluid was split were selected to be 50 μm in width, and it was
57
decided to use KOH etching to form them to reduce processing costs, thus creating
triangular channels (see Chapter 2 for a discussion of etching techniques). To minimize
DRIE etch time (and thus etch cost), as well as to minimize mixer volume, the focuser
and manifolds were etched to a depth of 250 μm. The mixing channel was designed to be
4.5-mm-long to ensure sufficient time for mixing.
The pressure drops along the manifolds, through the two sets of through-holes, within
the focusing cavity, and along the mixing channel were calculated using the HagenPoiseuille equation, with the equivalent diameter Navier-Stokes solution for rectangular
channels obtained from literature.70, 71 This was applicable to the focusing cavity because
its cross-section perpendicular to flow at any point is a rectangle; thus, its pressure drop
can be calculated by integration using the aforementioned formulae. For the triangular
distribution channels, the pressure drop was calculated using the solution for isosceles
triangular channels, as discussed in section 2.2.1 (equation 2.2).70, 72 The length of the
narrow channels was such that the pressure drop across them, when added to those of the
through-holes, the focuser, and the mixing channel, would be significantly greater than
the pressure drop across the distribution manifolds (the sections visible at the top of the
mixer images in Figure 3.1a and c, encompassing the arrays of flow through-holes). The
channels were made 7.5-mm-long, resulting in the manifolds having 0.6% of the pressure
drop of the subsequent mixer elements and satisfying the criterion. Overall, the mixer
was calculated to have a pressure drop of 0.57 bar/Qμ, where Q is the total flow through
the device in mL/min, and μ is the viscosity of said flow, in cP. The overall volume of
the mixer was 4.1 μL.
In addition to the distribution channels, the KOH etch also defined a shallow trench
on the back side beneath the mixing channel. This was intended to prevent a section of
borosilicate glass from bonding to the silicon, making it possible to snap off that glass
piece to expose the silicon surface.
This would allow for more rapid thermal
equilibration of the reactor with its ambient medium, which would permit direct reactor
cooling when exothermal mixing is involved.
58
3.2.2 Two-Wafer Stack
To improve upon the first-generation design, several modifications of the design were
made to further improve mixing, decrease pressure drop, simplify the fabrication
procedure, and expand the utility of the devices. The mixing was improved by increasing
the total number of lamina to 133 from 101, keeping the mixing channel width the same
at 500 μm. For two streams to be mixed, this decreased the mixing time by 73%.
To expand the utility of the devices, a design was made for a mixer of 3 streams, as
well. Many chemical syntheses require three reagent streams to be mixed for a reaction
to occur; therefore, having a single 3-stream device is more practical and streamlined
than two sequential 2-stream devices. The 3-stream device had the lamina of the three
streams, A, B, and C, stacked in the manner A-B-C-B-A-B-C-B-… This allows for rapid
mixing while maintaining a sequence of interactions, such as what may be necessary if
one reagent is an activator or an inhibitor. This is similar to a simple stacked on-chip
mixer used in previously developed devices.29
The largest pressure drop of the first-generation device, the distribution channels, was
necessary to evenly distribute the flow along the manifolds. Thus, if the pressure drops
of the manifolds were reduced, it would be possible to decrease the pressure drop of the
distribution channels. The manifold could be enlarged (thus reducing the pressure drop)
by removing it from the chip, and instead using a compression gasket to act as the
manifold.
The second-generation mixers were designed such that the through-holes for the flow
to enter the chip are exposed, relying on a gasket manifold to distribute the flow and to
provide a simple interface to the chip. This also significantly simplified the fabrication
process. Because there is no on-chip manifold, the distribution channels can be on the
same side of the wafer as the focuser cavity and the mixing channel. Therefore, only one
glass wafer is necessary, capping the channels and the focusing cavity. This eliminates
the necessity to process a glass wafer and reduces the number of bonding steps to one.
The manifold was designed to be produced out of elastomer gasket sheets, 1/16” thick
(1.6 mm). If compressed, the thickness may decrease down to 1 mm, or 4 times that of
the first-generation on-chip manifold. Thus, keeping other dimensions the same, the
pressure drop along the manifold is decreased by a factor of 44 = 256. This large
59
reduction allows for the distribution channels (which are kept 50 μm in width, as in the
previous design) to be etched by DRIE to the same depth as the main cavity, here
selected to be 200 μm. The ability to etch the channels identically to the focuser/mixer
removes one mask, one lithography step, and one etch step, greatly streamlining the
fabrication process.
At a minimum length of 3 mm and combined with the remaining one set of throughholes (while accounting for there being more channels than in the previous design), this
provides a total pressure drop approx. 200 times that of the manifold, satisfying the flow
distribution criterion. The overall pressure drop across the mixer was now 10-2 bar/Qμ,
where Q is the total flow through the device in mL/min, and μ is the viscosity of said
flow, in cP. This value is a factor of 50 lower than in the first generation. The overall
volume of the new mixer was 7.7 μL due to the wider focusing zone to accommodate
more lamina. The second-generation two-stream and three-stream mixers are shown in
Figure 3.2.
Figure 3.2. Two-stream (left) and three-stream (right) interdigitated silicon micromixers.
3.3
Micromixer Fabrication and Packaging
3.3.1 First-Generation Devices
The detailed process run sheet for the micromixer fabrication process is given in
Appendix A. In brief, the procedure for the fabrication consisted of nested mask layer
deposition, back-side lithography, back-side etching, two-step front-side lithography, two
front-side DRIE etches, protective layer growth or deposition, and two steps of bonding.
60
Figure 3.3 illustrates the three masks used to fabricate the silicon micromixer, and Figure
3.4 provides a schematic of the step-by-step fabrication process.
(a)
(b)
(c)
Figure 3.3. First-generation micromixer lithography masks: (a) Front-side manifold and mixing
focuser (the three circles designate the access holes through the top Pyrex layer); (b) backside
pressure drop channels (triangular, KOH-etched), and trench for silicon exposure; (c) through-holes.
(a)
(a) Oxidized silicon wafer with pressure-drop channels defined
in the oxide by buffered oxide etch.
(b)
(b) KOH-etched pressure drop channels (aligned into the
page), with mask oxide removed.
(c)
(c) Re-oxidized wafer with the manifold defined in the oxide,
and the through holes defined in photoresist (nested mask).
(d)
(d) Etched manifold and through holes by DRIE, and reoxidized wafer.
(e) Silicon wafer bonded to a Pyrex wafer on the bottom and to
a drilled Pyrex wafer on top.
Dark grey = silicon
Light grey = silicon oxide, including Pyrex for (e)
Black = photoresist
(e)
Figure 3.4. First-generation micromixer fabrication process.
Pyrex and silicon wafers were purchased from Silicon Quest International Inc., Santa
Clara, CA. Silicon wafers (6” diameter, 675 μm thick, (100) crystal surface) were
prepared by growing 200 nm of wet thermal oxide (20 min. at 1000ºC with H2/O2 flow,
MRL Industries Model 718 System Atmospheric Oxidation Furnace) to serve as the mask
61
layer for masks 1 and 2 in Figure 3.3. To define the back-side pressure drop channels
(mask 2), photolithography was performed using 1 μm of thin photoresist (OCG 825
positive resist), following the SOP using an Electronic Visions EV620 Mask Aligner. As
discussed in section 2.2.1, it is critical during the UV exposure to align the photomask to
the crystal plane of the wafer. Similar to the mask for KOH-etched microreactors in
Chapter 2, the mixer back-side mask has a number of closely spaced horizontal slits at the
region of the wafer flat for wafer alignment. The pressure-drop channels were defined in
the oxide layer using buffered oxide etch (BOE, 7:1 volume ratio of 40% NH4F in water
to 49% HF in water) and etched in silicon by potassium hydroxide (25 wt% KOH in
water at 80ºC for 30 min) to self-termination by <111> crystal planes.
Next, the manifolds, the focuser, and the mixing channel (mask 1) were
lithographically defined on the wafer front-side in the oxide layer, also using thin resist
and BOE to etch the oxide. Thick photoresist (AZ 9260) was then used, at 10-μm
thickness and following its respective SOP for coating, exposure, and development (with
AZ 440 developer), and the flow-through holes (mask 3) were defined. DRIE (ICP Deep
Trench Etching System, Surface Technology Systems, Newport, UK) was used to
vertically and anisotropically etch the flow-through holes to a depth of 450 μm, after
which, the thick resist was stripped by piranha in 10 minutes. The wafer was mounted
onto a handle wafer to prevent helium leak in DRIE, and the final etch of mask 1 and
mask 3 features was performed The oxide served as a hard mask for the DRIE etch of the
features of mask 2 to a depth of 250 μm.
The oxide layer was removed by HF, and a new layer of oxide was grown (500 nm,
200 minutes at aforementioned conditions) to serve as a relatively chemically inert layer,
functionally identical to laboratory glassware. For the top pieces, Pyrex wafers (6”
diameter, 650 μm thick) were outsourced for drilling of the holes (1/16” diameter) to
Bullen Ultrasonics Inc., Eaton, OH. The drilled wafers were anodically bonded to the
front-sides of Si wafers, and the back-sides were capped by anodically bonding an
unprocessed Pyrex wafer. During dicing, the backside glass was scored along the etched
trenches to allow it to be broken off easily, exposing the silicon surface for enhanced heat
transfer relative to a Pyrex-covered surface.
62
3.3.2 Second-Generation Devices
The detailed process run sheet for the second-generation process is given in Appendix
A. In brief, the procedure for the fabrication consisted of two-sided lithography, two
DRIE etch steps, protective layer growth or deposition, and one step of bonding.
The materials and equipment used in this process are identical to those used for the
first-generation devices, with several of the steps having been eliminated and simplified.
As discussed in section 2.2.1, two-sided lithography was performed prior to any etching.
Lithography and etching were performed on bare wafers without hard mask layers.
After both sides of the wafer were coated with thick resist (with the back-side coated
with 20 μm to compensate for the lack of an oxide layer), the focuser, mixing channel,
and pressure-drop channels (first mask) were defined on the front-side, and the flowthrough holes (second mask) were defined on the back-side. DRIE was used to etch the
front-side to a depth of 200 μm. The wafer was then mounted onto a handle wafer, and
flow-through holes were etched to a depth of 500 μm. A 500-nm layer of wet thermal
oxide was then grown, and the front-side was bonded to an unprocessed Pyrex wafer.
(a) Virgin silicon wafer with pressure-drop channels
and mixing cavity defined on the front-side and ports
to the channels defined on the backside, both in thick
photoresist.
(a)
(b) Etched 500-μm-deep through-holes by DRIE.
(b)
(c) Etched 200-μm-deep channels and mixing cavity
by DRIE, and oxidized the wafer.
(c)
(e) Bonded processed silicon wafer to a Pyrex wafer.
Dark grey = silicon
Light grey = silicon oxide, including Pyrex for (e)
Black = photoresist
(d)
Figure 3.5. Second-generation micromixer fabrication process.
63
3.3.3 Compression Packaging
The micromixers were packaged into compression chucks designed for standard ¼-28
flat-bottom nut-and-ferrule fittings (P-200 and P-201, Upchurch) for connection to fluid
tubing. For the first-generation mixers, only o-rings compression was achieved with
Teflon o-rings, size 005 (9559K113, McMaster). The chucks were machined out of
Hastelloy at the MIT Central Machine Shop. The materials were selected to provide
chemical compatibility with a wide range of chemicals, including strong acids and bases,
organics, and chlorinated solvents. The bottom pieces of the chucks were made of
polycarbonate to allow for visible access to the compression; the material selection was
not critical because this piece did not contact any of the fluid paths. The fittings are
easily connected and disconnected, allowing for the construction of a modular system
with easy probing of reaction conditions, such as pH, at various points along a reactor
chain if several such mixers are used in a process.
For the second-generation mixers, similar compression chucks were made. To reduce
the cost of the chucks, they were machined out of poly(ether ether ketone) (PEEK),
which has a fairy wide chemical compatibility, by Proto Labs (Maple Plain, MN). The
compression was achieved with Kalrez® materials for maximal chemical compatibility at
the seal while providing sufficient compressibility to allow the use of larger gaskets. A
size-005 o-ring (9568K33, McMaster) was used at the outlet, and custom-machined
manifold gaskets were applied at the inlet. The gaskets were machined from a Kalrez®
gasket sheet (CASS-.050X6X6-K7009, Acme Rubber, Tempe, AZ) using a water-jet
(OMAX® JetMachining® Center) available for student use at the MIT Hobbyshop. The
water-jet is capable of machining to very high tolerances, which is crucial because the
gasket must precisely fit within the chuck cavity to achieve compression. When the
gasket fits well, compression is achieved with very little force.
In addition to the fluidic connection for the micromixer, the 2nd-generation chuck also
provides a cavity and ports for cooling fluid. The ports are 10-32 threads designed for
barbed fittings (P-665, Upchurch) for hose connections.
The cavity is compressed
against the chip using a Viton® gasket, machined on the aforementioned water-jet from a
gasket sheet (86075K22, McMaster). This allows for direct cooling of the silicon side of
64
the chip at the mixing channel, which may be necessary for highly exothermal mixing.
Figure 3.6 shows the compression chuck and gaskets for the three-stream mixer.
(b)
(a)
(c)
(d)
Figure 3.6. Three-stream mixer compression chuck: (a) SolidWorks rendering; (b) chip side;
(c) fitting side; and (d) gaskets.
3.4
Micromixer Qualification
3.4.1 Micromixer Characterization Method
Many techniques exist for the characterization of mixing of various types and on
different scales. A current and comprehensive review of these techniques and their
applicabilities can be found in the literature.73 These methods typically consist of either
dilution-based (optical monitoring of colored or fluorescent species or outlet transecting
to quantify concentration gradients) or reaction-based (pH-indicating reactions, reactions
producing a colored or easily detectable species, or competing reactions based on the
above). Dilution-based characterization requires excellent visual access to the mixing
device, which is not available for compression-packaged silicon mixers, as they are not
fully transparent as are their glass or poly-(dimethylsiloxane) (PDMS) brethren. Thus,
reaction-based methods were selected, further narrowing it to ones with competing pHbased reactions to be able to study extremely fast mixing. This subcategory has several
methods, some of the better-known ones including the Bourne method for studying
micromixing in stirred tanks74 and the Villermaux/Dushman method,75-78 which is
frequently preferred due to its much greater ease of use. The latter has also been
frequently adapted for continuous flow applications.79-83
The mixing performance of the micromixers was evaluated by using the
Villermaux/Dushman method, as adapted for continuous flow applications by Panić et
65
al.83 This method takes advantage of two competing parallel reactions (Scheme 3.1).
Under acidic conditions, potassium iodide reacts with potassium iodate to form iodine in
a fast reaction. This reaction competes for the acidic protons with the near-instantaneous
neutralization reaction by the borate-based buffer.
Scheme 3.1. Competing parallel reactions of the Villermaux/Dushman method.
3.4.2 Characterization Experimental Setup
The experiments involved a solution of KI and KIO3, buffered with NaOH and boric
acid, mixed with dilute sulfuric acid.
If mixing occurs rapidly, the acid is nearly
instantaneously fully neutralized by the basic H2BO3- ions. However, when mixing is
poor, the gradients in [H+] permit local excess of protons, which react with iodide and
iodate in a comproportionation reaction, forming I2. It then further reacts with the excess
iodide in solution to form a strongly UV-absorbent triiodide ion with characteristic
absorption bands at 286 nm and 353 nm. Thus, the mixing performance is inversely
related to the amount to triiodide detected.
Reagents were used as received and prepared in HPLC-grade water purchased from
VWR. Analytical reagent grade sulfuric acid, sodium hydroxide, and boric acid were
supplied by Mallinckrodt Chemicals (Phillipsburg, NJ). Potassium iodide was purchased
from Acros Organics (Fairlawn, NJ), and potassium iodate, from EMD Chemicals in San
Diego, CA. Using the concentrations given by Panić et al.,83 two solutions were prepared.
Sulfuric acid was prepared at 0.015 M. The buffered KI/KIO3 solution was prepared by
separately preparing solutions of KI (0.0319 M) and KIO3 (0.00635 M), both containing
0.0909 M each of NaOH and H3BO3. Equal volumes of these two solutions were mixed
immediately prior to the experiment. The sulfuric acid and iodide/iodate solutions were
then degassed by ultrasonication and pumped through the micromixer using two Harvard
Apparatus PHD 2200 syringe pumps set to identical flow rates.
66
The reagents were delivered by 10 mL Hamilton Gastight® syringes (model 1010).
These syringes were fitted with Teflon® Luer locks (Upchurch P-628) and connected to
1/16” OD 0.03” ID Teflon® inlet tubing by Tefzel® nut/ferrule fittings (Upchurch P-245
and P-200N). Identical nut/ferrule fittings connected the inlet tubing, as well as the outlet
tubing, to the compression chuck inlets. For the three-stream micromixer, one solution
was split between two syringes and flowed into the two outside inlets (A and C, per the
notation in section 3.2.2) at one-quarter of the total flow rate each, while the other
solution was flowed into the central inlet (B, per the same notation) at one-half of the
total flow rate. Thus, each lamina was surrounded on both sides by lamina of a different
solution. Equivalent results were found when either the acid or the iodide/iodate solution
was split into flows A and C.
The outlet of the fluidic chuck was 5 cm of 1/16” OD 0.01” ID Teflon® inlet tubing
and a PEEK single-piece HPLC fitting (Upchurch F-130). Based on the results by Panić
et al.,83 under the chosen experimental conditions, the length and volume of the outlet
tubing does not influence the measurement of triiodide because all of the acid is
completely consumed prior to reaching the outlet.
Figure 3.7. Villermaux/Dushman method experimental setup schematic, taken from Panić et al.83
The outlet was connected directly into the inlet of a Waters 2996 Photodiode Array
Detector (PDA). The outlet of the PDA was connected to an inline 40-psi backpressure
regulator (Upchurch P-785), which ensured stable and steady flow through the PDA flow
cell. The inline PDA allowed for real-time online monitoring and detection of triiodide,
with a spectrum sampling rate of 1.0 Hz. Each reported absorbance value represents an
average of at least 10 s of measurement after attaining steady state, which was gauged
67
visually by observing the real-time spectrum reported by Waters Empower® software.
The mixing performance of the micromixers was measured at several total volume flow
rates in the range of 40 to 2000 μL/min. All experiments were conducted at room
temperature. Outlet pH was constantly in the range of 9 to 10, as measured by either EM
Science colorpHast® indicator strips or Baker-pHIX pH papers, confirming accurate
concentrations of reagents.
3.4.3 Characterization Results and Discussion
Figure 3.8 depicts the mixing performance and the mixing efficiency of the silicon
micromixers, as measured by the absorbance of triiodide at 286 nm, with flow rates at the
conditions described in the Experimental section and pressure drops calculated at
corresponding flow rates. Lower absorbance indicates that less triiodide was formed,
which implies better mixing. The figure does not display the data for the secondgeneration two-stream mixer because its mixing performance was very similar to that of
the first-generation mixer. Its performance was significantly better due to the much
lower pressure drop.
The micromixer performance is compared to that of a commercially available glass
micromixer (mgt mikroglas, technik AG, Mainz, Germany) that has 31 lamina (15 and 16
for each of the two flows) 50 μm wide and 150 μm deep. The data for the performance
of the mikroglas mixer at the identical conditions was obtained by Panić et al.;83 no error
bars were available for said data.
At flow rates of 40 to 250 μL/min, the two-stream micromixer shows a very similar
performance to the commercial mixer.
With increasing flow rate, the mixing
performance improves for both mixers because the liquid spends less time in regions of
larger diffusion length (beginning of the focuser cavity); thus, the onset of mixing occurs
more rapidly, preventing the competing reaction. The three-stream mixer shows a similar
performance at flow rates at and above 250 μL/min; however, its performance remains
very high down to the lowest tested flow rate, showing optimal performance for mixing
two fluids.
68
Absorbance of triiodide at 286 nm
0.5
(a)
0.4
1st-gen. 2-stream mixer
0.3
2nd-gen. 3-stream mixer
Commercial micromixer
0.2
0.1
0
0
500
1000
1500
Total Flow Rate (μL/min)
2000
Absorbance of triiodide at 286 nm
0.5
2500
(b)
0.4
1st-gen. 2-stream mixer
0.3
2nd-gen. 3-stream mixer
Commercial micromixer
0.2
0.1
0
0
0.2
0.4
0.6
0.8
1
Pressure drop (bar)
1.2
1.4
Figure 3.8. Micromixer performance (a) and efficiency (b). Blue diamonds correspond to the firstgeneration silicon micromixer; green squares correspond to the second-generation 3-stream silicon
micromixer; red triangles correspond to a single channel in PDMS; filled black circles correspond to
a commercial glass micromixer from mgt mikroglas, technik AG, Mainz, Germany, as reported by
Panić et al.83 Error bars are shown within the open data symbols; no error bars were available for
the glass micromixer.
69
The efficiencies of the first-generation mixer and the mikroglas mixer appear to lie on
the same curve, showing that they deliver similar mixing performance at a given pressure
drop. By comparison, the second-generation three-stream mixer (as well as the twostream mixer, not shown on the graph) delivers superior performance at a mere fraction
of the pressure drop.
At the conditions used, mixing can considered good if the absorbance of triiodide is
below 0.5. A comparison was made where the two solutions described in the above
section were mixed in batch. The acid solution was poured over a period of 10 seconds
into an equal volume of the iodide/iodate solution (10 mL of each) in a 20 mL vial
equipped with a magnetic stirbar. The resulting solution was yellow in color, and when
run through the UV flow cell, it saturated the detector (absorbance value above 5) at 286
nm. Thus, flow through a micromixer results in superior mixing.
3.4.4 Cooling of Three-Stream Micromixer
The micromixers have been applied in the setup of continuous-flow synthesis of an
active pharmaceutical ingredient (API) (Aliskiren, Novartis-MIT Center for Continuous
Manufacturing). In one of the steps of the synthesis, an acidic HCl solution in a mixture
of water and ethyl acetate will be quenched with NaOH and additional ethyl acetate in a
three-stream mixer. This setup is designed to ensure rapid quenching of HCl while
providing simultaneous extraction of one of the reaction intermediates from the aqueous
into the organic phase.
To experimentally verify whether the micromixers are capable of performing the
neutralization at the required flow rates, an experimental simulation of the process step
was performed using the three-stream micromixer packaged in a compression chuck that
contained a flow path for coolant to contact the silicon device, as shown in Figure 3.6.
The PEEK chuck and Kalrez® gaskets provide chemical compatibility with the acidic,
basic, and organic streams. The coolant flow was provided using a recirculating chiller
(Fisher Scientific Isotemp® Refrigerated Bath, #13-874-9), connected by hoses to the
barb adapters in the chuck (see section 3.3.3) to remove some of the heat generated by the
acid/base neutralization and thus prevent the etching of the silicon by the basic stream.
70
All chemicals were purchased from VWR as reagent-grade and used as received. The
fittings, tubing, and syringe pumps were identical to those used in section 3.4.2. The
syringes used were 60 mL in volume, with Luer lock tips (309653, Becton Dickinson). A
stream of 6 M HCl in 50:50 ethyl acetate/water (representing the stream containing the
reaction intermediate) was mixed with an equal flow rate of pure ethyl acetate and a
stream of 4 M NaOH at a flow rate sufficient to result in a slightly basic pH (monitored
by EM Science colorpHast® indicator strips). A flow rate of 6 mL/min of the acidic
stream was reached using Harvard syringe pumps, which is representative of the flow rate
of the corresponding acidic stream in the planned synthesis process at a production level
of 60 g/hr of the API. The total flow rate through the micromixer was 30 mL/min, with
no undue strain observed on the syringe pumps or the plastic syringes. At these flow
rates, with a flow of coolant set at 13ºC, the temperature of the exiting stream directly
following the mixer was 39ºC, reaching 54ºC without coolant flow. Based on the heat
capacities and densities of brine (formed with HCl/NaOH neutralization) and ethyl
acetate, the maximal energy removal rate was calculated to be 25 W. No damage was
observed to the mixer following an extended reaction period of 45 minutes.
3.5
Conclusion
A set of silicon micromixers has been developed to enable rapid mixing of two to
three homogeneous liquid streams within 10 milliseconds.
The mixers have been
designed to be simple to microfabricate and to have a very low pressure drop of 10-2 bar
per mL/min flow of 1-cP liquid, thus providing much higher mixing efficiency than
available commercial micromixers. The mixer performance has been verified using the
established flow-based Villermaux/Dushman qualification method and has been shown to
provide excellent mixing in the tested total flow range from 40 to 2000 μL/min, with
mixing improving at higher flow rates. The micromixers have been applied to flows of
up to 30 mL/min. Additionally, the packaging method provides for direct access of a
cooling liquid stream to the silicon surface, thus enabling highly exothermal mixing to be
performed in these devices.
71
Chapter 4.
Sodium Nitrotetrazolate Kinetics†
To demonstrate the utility of microfluidics in enabling safe and efficient kinetic
studies of rapid reactions with hazardous intermediates and/or products, the kinetics of
the two-step synthesis of sodium nitrotetrazolate via a diazonium intermediate was
studied. The ability to directly synthesize sodium nitrotetrazolate in a safe manner via
only the involved diazotization and Sandmeyer reaction steps is of great interest due to
the hazards typically associated with this process and the inefficiencies involved.
Additionally, a full understanding of the kinetics of these reactions would allow the
efficient performance of this synthesis. This chemistry is highly sensitive to pH and ionic
strength, and a model incorporating these elements is necessary.
The intermediate of this synthesis is highly energetic and shock-sensitive in its dry
crystalline form. Thus, it is desirable to minimize the amount of intermediate present at
any given time and to prevent its crystallization, both of which are enabled by continuous
flow and microfluidics.
Additionally, the high sensitivity to pH and, for the
diazotization, the very fast reaction rate make extremely rapid mixing imperative for
accurate kinetic studies.
The first-generation micromixers developed in Chapter 2 were used to safely perform
a quantitative kinetic study of the direct two-step synthesis of sodium nitrotetrazolate.
Orders of reactions and temperature dependence of both steps as well as pH and ionic
strength dependence of the second step were evaluated. Scale-up of the reaction to
industrial levels was also demonstrated, as successful production of 4.4 g/hour of NaNT
in solution was ultimately achieved in a compact footprint using the kinetic data.57, 58
†
This chapter describes work done in close collaboration with Edward R. Murphy and
Jason G. Kralj, who at the time were fellow doctoral students in the laboratory of Prof.
Klavs F. Jensen.
72
4.1
Motivation
The ability to perform on-demand synthesis of hazardous intermediates is one of the
more promising uses of microreactors, along with the opportunity to gain mechanistic
understanding and rate parameters for scale-up to production levels.17, 84 With reduced
thermal mass and rapid mixing, conditions within a microreactor can be more tightly
controlled than those in a traditional apparatus.18 Furthermore, the use of a microreactor
limits the quantity of reactive intermediate to only that required for immediate
processing, thus simplifying containment in the event of a reactor failure. The enhanced
heat and mass transfer within microreactors also allows faster equilibration to steady state
and shorter time between experiments, thus reducing reagent consumption and
accelerating reaction optimization, and grants access to reaction conditions that would be
impractical to pursue by standard laboratory techniques.28, 85
Reactions involving diazonium intermediates, such as the synthesis of azo dyes, have
been of great interest in the microreactor community for some time as an example of
multi-step synthesis the microscale safety advantages. Reactive intermediates such as
tetrazolediazonium are extensively used in many chemical industries, including
medicine,86,
87
biology,88,
89
and explosives.90,
91
However, the processes are often not
well understood or optimized due to the difficulty in obtaining reaction kinetics and in
scaling up from laboratory to pilot to production levels. The difficulties are presented by
the instability of the reactive intermediates, making typical kinetic studies both difficult
and potentially highly dangerous, requiring numerous precautions. These reactions have
been performed safely using monolithic micro- and nanoreactors.64,
92
Additionally,
combinatorial synthesis of azo dyes in immiscible liquid slugs has also been
demonstrated.93
To further advance the study of chemical synthesis via diazonium intermediates, a
system of modular mixers with adjustable residence volumes for the reaction was used,
applying the first-generation rapid micromixers discussed in Chapter 2 and using polymer
tubing to provide the residence time for the reaction steps. Such a system permits
isolating intermediate species and varying the ratio of reaction times among reaction
steps. Specifically, the system enabled exploring the reactions involved by separately
analyzing the initial diazotization step, as well as characterizing the effects of reaction
73
conditions such as temperature and pH on conversion and selectivity. Moreover, the
micromixer devices accommodated higher flow rates than typically possible in fixed
volume microreactors, thus enabling the use of larger volumes and increased production.
4.2
Sodium Nitrotetrazolate Synthesis Description
The target product sodium nitrotetrazolate 3 (NaNT) was selected to demonstrate a
safe and efficient method of both performing kinetic studies and synthesizing a
potentially, highly explosive compound.
NaNT 3 is a precursor for the commercial
product tetraamine-cis-bis(5-nitro-2H-tetrazolato-N2) cobalt(III) perchlorate (BNCP).94
It is directly synthesized when 5-aminotetrazole 1 (AT) reacts with nitrous acid to
produce the 5-diazonium-1H-tetrazole 2 (DHT) intermediate, which further reacts with
the nitrite ion via a modified Sandmeyer reaction (Scheme 4.1).
1 N
NH
NaNO2
NO2
N
H+
N
3 N+
NH2
N
N
2
N
NO 2-
N
NH
-N 2
N
N
N
-N
Na+
OH
H+
OH-
N
N
N
N
NH
4
N
Scheme 4.1. Two-step formation of sodium nitrotetrazolate 3 (NaNT) from 5-aminotetrazole 1 (AT)
via 5-diazonium-1H-tetrazole 2 (DHT) intermediate. The acid-base equilibrium between DHT 2 and
the non-reactive 5-hydroxydiazonium-1H-tetrazole (HDHT) 4 is also shown.
In industrial processes, batch vessels are typically employed for the synthesis, and the
direct synthesis scheme is avoided due to the hazards associated with it. Both DHT 2 and
NaNT 3 are highly unstable in their crystalline form and, to an extent, in increased
concentrations (6-7% for DHT 2).95, 96 N2 gas released in the second reaction step (see
Scheme 4.1) tends to create froth in a stirred batch vessel. This froth can accumulate on
74
batch vessel side walls and become concentrated or even form dry solids, leading to
potentially explosive conditions.
Therefore, commercial production of NaNT 3 has
typically applied alternative syntheses such as the use of copper(II) sulfate (CuSO4) that
stabilizes DHT 2 and complexes with NaNT 3.
However, this approach has the
disadvantages of requiring elevated temperatures (70ºC) and addition of base to release
NaNT 3 in its non-stabilized form for further reaction, causing some degradation of
NaNT 3, reducing reaction yields, and forming solid copper oxide (CuO) precipitation.9799
Furthermore, both reaction steps are highly pH sensitive, as are the intermediate and
the product. DHT 2 degrades with time at low pH (such as the range at which it must be
synthesized).95, 97 In addition, at high pH, DHT 2 instead takes the form of non-reactive
5-hydroxydiazonium-1H-tetrazole (HDHT) 4 (Scheme 4.1). Thus, NaNT 3 must be
synthesized at a mildly acidic range, requiring introduction of a buffer and/or base.
However, the amount of buffer/base must be carefully controlled, as NaNT also degrades
at high pH and elevated temperatures.
Because the reaction is exothermic, local
temperature and concentration gradients can cause product loss, leading to mixing being
a crucial parameter during both production (to maximize yield) and kinetic study (to
ensure accurate study of actual reaction). With these considerations, a continuous flow
system incorporating micromixers was developed, allowing for both rapid mixing and
precise time control of composition with very little effort.
4.3
Micromixer-Based System Design
4.3.1 DHT 2 Formation Study
The kinetics of the first step of direct NaNT 3 synthesis, the production of DHT 2,
were evaluated by mixing a stream of 5-AT 1 in dilute sulfuric acid with a stream of
excess NaNO2 in a micromixer and evaluating the quenched effluent. As the reaction can
only proceed under acidic conditions, NaOH was used to quench the reaction, with the
reaction and quench streams mixing in a second micromixer to ensure very rapid
quenching (Figure 4.1).
75
Figure 4.1. Reaction setup for kinetic study of DHT 2 formation.
Reagents were used as received and prepared in deionized water that was filtered
through a Millipore Academic Milli-Q water purifier. 5-Aminotetrazole 1 was purchased
from Lancaster Synthesis, Inc. in Pelham, NH. Analytical reagent-grade sulfuric acid and
sodium hydroxide were supplied by Mallinckrodt Chemicals (Phillipsburg, NJ). Sodium
nitrite (97%) was purchased from Alfa Aesar in Ward Hill, MA. Three solutions were
made for each experiment: a solution of 5-AT 1 (0.025-0.05 M) in 1.5 M sulfuric acid, an
aqueous solution of sodium nitrite (0.025-0.05 M), and a solution of 4 M sodium
hydroxide to quench the reaction. The solutions were degassed by ultrasonication prior
to experiments.
The reagents were delivered to the micromixers using the same setup and equipment
as for the micromixer qualification (section 3.3.2).
A single multi-head Harvard
Apparatus PHD 2200 syringe pump was used with all three syringes, delivering equal
flow rates of each solution. The tubing connecting syringes to micromixers was 0.04”
ID. After mixing the 5-AT 1 and NaNO2 streams in the first micromixer, the reaction
stream entered the reaction zone consisting of a 17.5 cm length of 0.020” ID Teflon®
tubing, providing 39.5 μL of reaction volume (including 4.1 μL of the micromixer
volume).
Because the total residence volume was constant, reaction times were
controlled by varying the flow rates of the syringes. The total reaction flow rate was
varied from 240 to 2000 µL/min, corresponding to residence times of 9.9 seconds to 1.2
seconds, respectively. The reaction zone tubing connected to the second micromixer,
where the reaction stream mixed with the NaOH quench stream (at a volumetric flow
76
ratio 2:1) and exited through an additional 5 cm piece of 0.04” ID tubing. Residence
times from 1.32 s to 9.8 s were investigated, controlled by adjusting the flow rates by the
syringe pump.
Reaction temperature control was achieved by submerging both compressed
microreactors and the reaction zone tubing completely into an ethylene-glycol-filled
heater/chiller recirculating bath (Neslab Endocal). To evaluate the reaction temperature
dependence, the reaction was performed at four temperatures between 5ºC and 28ºC,
inclusive, with 1.32 second residence time. For investigation of reaction order, the
reaction was performed without the bath at room temperature, which was measured to be
21 ± 1ºC.
Reaction samples were collected into two-dram glass vials, with at least three samples
collected per set of experimental conditions. At least five residence times were allowed
to pass between attaining new experimental conditions and sample collection. Collected
samples were diluted by a factor of 10 with mobile phase, loaded into a Waters 717+
Autosampler, and analyzed using a Waters Nova-pak© C18 column (3.9 x 150 mm). The
mobile phase was an aqueous solution of 0.1 M monobasic phosphate buffer pumped
isocratically at 1 mL/min by a Waters 1525 binary pump. The eluent was monitored on a
Waters 2996 Photodiode Array Detector. Elution of all compounds was complete after 5
minutes. Because of the instability of DHT 2, the reaction conversion was measured
indirectly by measuring the concentration of HDHT 4, which is in equilibrium with the
diazonium salt. The excess sodium hydroxide in the quench ensured a quantitative
conversion to HDHT 4 and, as a result, no NaNT 3 production was observed in this
portion of the study. Peak absorbance of NO2- was observed at 209.7 ± 0.5 nm, of 5-AT
1, at 217.9 ± 0.5 nm, and of HDHT 4 at 261.5 ± 0.5 nm.
4.3.2 NaNT 3 Formation Study
The kinetics of the second step of direct NaNT 3 synthesis, producing the desired
NaNT 3, were evaluated by mixing the unquenched product stream of the DHT 2
production setup with a buffer solution in a micromixer and evaluating the quenched
effluent. As the reaction can only proceed in a pH range from mildly acidic to very
mildly basic, NaOH was used to quench the reaction. Because this reaction step is
77
significantly slower than the first step, it was not necessary to quench the reaction in a
micromixer, as mixing through a T-mixer was sufficiently rapid for the reaction and
quench streams.
Reagents were used as received and prepared in deionized water that was filtered
through a Millipore Academic Milli-Q water purifier. In addition to the reagents used for
DHT 2 synthesis, sodium acetate (Mallinckrodt Chemicals, Phillipsburg, NJ) and sodium
phosphate (99%, EM Science, Gardena, CA) were used for the buffer solutions. Four
solutions were made for each experiment: a solution of 5-AT 1 (0.05-0.10 M) in 1.5 M
sulfuric acid, an aqueous solution of sodium nitrite (0.20-0.35 M), a solution of buffered
base, and a solution of 4 M sodium hydroxide to quench the reaction. The solutions of 5AT 1, NaNO2, and the quench were flowed at equal volumetric rates. The solutions were
degassed by ultrasonication prior to experiments. A buffered base solution required for a
pH of 5, comprising NaOH and sodium acetate (NaOAc) was used for studies of reaction
order and temperature dependence.
The composition of the solution of buffered base varied based on the desired pH
range of the reaction, assuming total conversion of 5-AT 1 to DHT 2 prior to mixing with
the buffer. The calculation of pH range was bounded at 0% conversion and at 100%
conversion of DHT 2 to NaNT 3, considering 2 mol H+ evolution per 1 mol of NaNT 3
generated. The values of pKa of nitrous acid, sulfuric acid (second deprotonation), acetic
acid, and phosphoric acid (all three deprotonations) were obtained from standard
handbooks. The pKa of 5-nitrotetrazole, the protonated form of NaNT 3, is -0.82.100 The
pKa of DHT 2 was approximated by titration of a product solution of DHT 2 of known
composition and was found to be approximately 8. However, the calculations of pH were
very insensitive to the pKa of DHT 2, given the low concentrations used and the nearly
neutral nature of DHT 2.
The calculations took as the input the intended input
concentrations of the reagents, 5-AT 1 and NaNO2, assumed 1.5 M sulfuric acid solution
of NaNO2 input and desired flow ratios of the three syringes (two reagents and buffer),
and the desired pH at the start of the reaction.
The outputs were the required
concentrations of NaOH and either NaOAc or sodium phosphate (NaPhos) to yield the
desired pH at the start of the reaction and that pH minus 0.15 at full conversion of DHT 2
to NaNT 3.
78
NaOAc was used for pH of and below 6, with concentrations of NaOAc and NaOH
varying from 0.6 M to 2.4 M and from 0 M to 3 M, respectively, in the syringe. The
buffered base solution was flowed at an equal flow rate as each of the other syringes.
NaPhos was used for pH of and above 6, with concentrations of NaPhos and NaOH
varying from 0.044 M to 1.2 M and from 1.5 M to 1.8 M, respectively, in the syringe.
The buffered base solution was flowed at a flow rate twice that of any of the other
syringes. This was done because the solubility of NaPhos was too low to use a more
concentrated buffered base solution at flow rates equivalent to those of other reagents.
The reagents were delivered using an expansion of the setup used for DHT 2 kinetics
analysis. For experiments using NaOAc buffered solution, a single multi-head Harvard
Apparatus PHD 2200 syringe pump was used with all four syringes, delivering equal
flow rates of each solution. A second pump was used for experiments with NaPhos
buffered solution, delivering the buffered base at twice the flow rate as any of the other
syringes. The tubing connecting syringes to micromixers was 0.04” ID. After mixing the
5-AT 1 and NaNO2 streams in the first micromixer, the reaction stream entered the
reaction zone consisting of a 29.6 cm length of 0.040” ID Teflon® tubing, providing 244
μL of reaction volume (including 4.1 μL of the micromixer volume). This volume was
sufficient to ensure complete conversion at the evaluated flow rates, which were varied
from 100 to 300 μL/min of each component during reaction order evaluation, providing a
residence time of 74 to 25 seconds, respectively, in the DHT 2 formation zone. This was
confirmed by analysis of reaction samples, which showed no presence of 5-AT 1.
As the second step of the reaction evolves nitrogen gas (Scheme 4.1), a vacuum
degasser was used to ensure no gas bubbles evolved during the experiments, allowing for
accurate residence time measurement. The vacuum degasser consisted of a machined
aluminum vacuum chamber with gas-permeable Teflon® AF tubing coiled within,
connected to a vacuum pump (1/2 HP, Leland Faraday M291 A) via a rubber vacuum
hose (Figure 4.2). No gas bubbles were seen entering or forming in the reaction zone
tubing during experiments. However, if the pump was turned off and the reaction flows
were allowed to proceed normally, evolution of gas slugs was seen within the Teflon® AF
tubing.101
79
Figure 4.2. Schematic and photograph of Teflon® AF based degasser, taken from Sahoo.101
The Teflon® AF tubing (0.036” ID, 48 cm, 361 μL) was used as the residence volume
for the conversion of DHT 2 to NaNT 3, along with the micromixer (4.1 μL) and small
pieces of tubing connecting the micromixer to the degasser and the degasser to the
quench T-mixer, for a total residence volume of 380 μL.
For reaction order
determination, reaction times were controlled by varying the flow rates of the syringes.
The total reaction flow rate was varied from 300 to 900 µL/min, corresponding to
residence times of 76 seconds to 25 seconds, respectively.
Upchurch NanoTight® PEEK tubing sleeves were inserted into the degasser ports, and
the gas-permeable tubing was threaded into them to a sufficient distance as to be held in
place at the port by an Upchurch NanoTight® headless fitting and ferrule (F-333N). The
second end of each sleeve was attached by another such fitting to an Upchurch
VacuTight® union (P-845-01), which, via a VacuTight® short fitting and ferrule (P-844)
and a short piece of 0.040” ID Teflon tubing, were attached to the micromixer chuck on
one end and to an Upchurch PEEK tee union (P-712) on the other end. At the tee union,
the reaction stream mixed with the NaOH quench stream and exited through an additional
10 cm piece of 0.04” ID tubing. The reversible fittings at the mixer chuck and tee union
allowed the mixer to be disconnected from the residence zone and the residence zone
from the quench. This permitted to measure the pH of the reacting solution before and
after the reaction with either EM Science colorpHast® indicator strips or Baker-pHIX pH
papers before reconnecting the quench to collect samples for HPLC analysis. The setup
schematic for NaNT 3 kinetic study is shown in Figure 4.3.
80
Figure 4.3. Reaction setup for kinetic study of the direct NaNT 3 synthesis.
Reaction temperature control was achieved by submerging both of the compressed
microreactors, the first reaction zone tubing, and the vacuum degasser completely into an
ethylene-glycol-filled heater/chiller recirculating bath (Neslab Endocal). To evaluate the
reaction temperature dependence, the reaction was performed at four temperatures
between 6ºC and 29ºC, inclusive, with 3 flow rates evaluated at each temperature. For
investigation of reaction order, the reaction was performed without the bath at room
temperature, which was measured to be 21 ± 1ºC. To measure temperature within the
chamber during temperature dependence experiments, a digital thermometer (Omega
HH-21A, with a K-type wire thermocouple) was threaded into the chamber through a slit
in the vacuum tubing, which was sealed with duct tape.
Reaction samples were collected into 2-dram glass vials, with at least three samples
collected per set of experimental conditions. At least five residence times were allowed
to pass between attaining new experimental conditions and sample collection. After
quenching, samples were collected, diluted, and analyzed by the same HPLC equipment
and method as that used for DHT 2 kinetics analysis. Peak absorbance of NaNT 3 was
observed at 256.7 ± 0.5 nm.
81
4.3.3 Scale-up to NaNT 3 production
The scale-up of direct NaNT 3 synthesis was performed on a system nearly identical
to the one used for the NaNT 3 kinetics evaluation, using identical reagents and
evaluation methods, diluting samples by a factor of 100 for analysis. Two sets of
experiments were performed.
In one, reaction concentrations and conditions were
selected based on determined kinetics to attempt to maximize production of NaNT 3.
The other set was performed at suboptimal conditions to demonstrate the sensitivity of
the reaction to pH at various temperatures.
Four solutions were made for each experiment: a solution of 0.4 M 5-AT 1 in sulfuric
acid (1.5 and 1.8 M for suboptimal and optimal sets, respectively), an aqueous solution of
1.6 M sodium nitrite, a solution of buffered base (NaOAc and NaOH), and a solution of 4
M sodium hydroxide to quench the reaction. For the optimal case experiments, more
acidic solution of 5-AT 1 was used to counterbalance the increased concentration of
NaNO2 in the first reaction. The buffered base and the quench were flowed at 0.9 times
the volumetric rates of the two reagents, and the buffered base solution was calculated to
provide a pH of 5 to 4 (from start to end of reaction) for the optimal case. A subset of
these experiments was performed without an inline quench, instead collecting 1.5 mL
samples and quenching them with 0.5 mL of the 0.4 M NaOH solution offline. For the
suboptimal case experiments, the base solution and quench were each flowed at 0.8 times
the volumetric rates of the reagent solutions, and the base solution was calculated to
provide between pH between 8.5 and 4.2, with a sharp titration from 7.5 to 5.2. The
solutions were degassed by ultrasonication prior to experiments.
For these experiments, the setup from the kinetic study of NaNT 3 was modified as
follows. Plastic single-use 60-mL Becton-Dickinson (BD) syringes were used to deliver
the reagents. To ensure the syringes did not yield when pumping, the flow rate was
verified by collecting the output in a 10-mL graduated cylinder for a set period of time at
the beginning of each experimental set and after changing flow rates. To accommodate
the higher pressures, two syringe pumps (identical to those in previous sections) were
used, with the two reagent streams on one pump, and the base and quench streams on the
second pump. The nitration step was performed in a 28-cm-long piece of ¼” OD, 0.188”
ID Teflon tube (5 mL residence volume) with no degassing. The ¼” OD tubing was
82
connected via a stainless steel Swagelok reducing union (SS-400-6-1) to short pieces of
1/16” OD, 0.04” ID Teflon tubing, which, in turn, were connected as previously
described to the second micromixer chuck and the quench tee. Degassing was initially
attempted, but the residence volume of 0.036” ID Teflon® AF tubing necessary for these
experiments created excessive pressure drop, making it highly impractical. Thus, the
residence time was not known for these experiments, as very large quantities of gas were
generated, creating a highly non-linear dependence of residence time on reagent flow
rate. During temperature evaluation experiments, the thermocouple was submerged into
the ethylene glycol bath along with the reaction tubing to confirm temperature at the
outer surface of the tubing.
No 5-AT 1 was observed in any of the experiments,
indicating full conversion of 5-AT 1 to DHT 2 in all cases.
4.4
Reaction Kinetics Evaluation and Scale-up
4.4.1 Kinetic evaluation of DHT 2 formation
The order of the reaction of DHT 2 formation from 5-AT 1 and HONO was
determined by analyzing the results of varying reagent concentrations (Figure 4.4). The
increase of UV absorbance of HDHT 4 linearly correlated to the decrease of absorbance
of nitrite, and the measured changes in concentration of 5-AT 1 corresponded nearly
identically to the measured changes in concentration of NO2-.
Combined with the
observation of no other peaks in the UV analysis, the result indicates that only the desired
reaction occurred in these studies.
Because the HPLC analysis is performed in a buffered medium, it could not be used
to determine the ratio of HONO to NO2- in the reaction stream. However, at the used
concentration of sulfuric acid (0.75 M in reaction stream), all of NO2- is assumed to be in
the dissociated form, based on its pKa of 3.35. The initial rate of reaction was
approximated by measuring the change in nitrite concentration from its starting value to
that at 1.2 seconds into the reaction.
For the baseline experiment with initial
concentrations of 0.0125 M each of 5-AT 1 and of NO2-, the initial rate was measured to
be 3.82×10-3 mol L-1 s-1. When the initial concentration of 5-AT 1 was doubled to 0.025
M, keeping the nitrite concentration the same, the initial rate was measured to be
83
3.82×10-3 mol L-1 s-1, or unchanged from the baseline case, and the conversion with time
followed an identical trend to the baseline experiment. When the initial concentration of
nitrite was also doubled (both 5-AT 1 and nitrite initially at 0.025 M) the initial rate
increased to 1.75×10-2 mol L-1 s-1, slightly over 4 times that of the baseline experiment,
and the conversion with time followed a trend that indicated a second-order reaction
when compared to the baseline case.
Combined, these results indicate that, at the
evaluated conditions, the reaction is zero-order with respect to 5-AT 1 and second-order
with respect to HONO, or following the rate law:
rDHT = k1[HONO]2
4.1
Figure 4.4. DHT 2 reaction order determination through consumption of nitrite (diamonds
correspond to initial concentrations of nitride and 5-AT of 0.0125 M each; triangles correspond to
the initial concentration of nitride of 0.0125 M and 5-AT of 0.025 M; X’s correspond to initial
concentrations of nitride and 5-AT of 0.025 M each; ---- represents the initial rate slope of the first
two data series, and ....... represents the initial rate slope of the third series).
The above result is somewhat surprising based on the known kinetics of diazotization
at moderate acidities. For the identical reaction using arylamine in place of 5-AT 1, at
acidities of 0.1 – 6.5 M [H+], the rate has been reported as the following equation:102
rDiaz = k1[ArNH 2 ][HONO]h0 + k2 [ArNH3+ ][HONO]h0
4.2
where h0 is the Hammet acidity function, which at this pH range is nearly equal to [H+].
However, for the same reaction at low acidities (pH > 2), the observed rate with
arylamine is equivalent to equation 4.1.103 This change in mechanism is caused by the
84
rate-limiting step becoming the formation of N2O3 from HONO, which is believed to be
the actual reactive species.104 In the case of 5-AT 1 as the reagent, it is possible that,
because tetrazole is more reactive than benzene due to the electron-donating effects of the
alpha nitrogen, the formation of N2O3 is the rate-limiting step even at higher acidities.
4.0
-1
-1
ln k1 (M s )
3.5
3.0
Ea/R = -4337 K
2.5
2.0
3.3×10-3
3.3E-03
3.4×10-3
3.4E-03
-3
3.5×10
3.5E-03
3.6×10-3
3.6E-03
-1
Inverse Temperature (K )
Figure 4.5. Arrhenius correlation between temperature and DHT 2 production rate constant.
Figure 4.5 shows the temperature dependence of DHT 2 formation as a plot of ln(k1)
vs. 1/T. The Arrhenius parameters were determined to be:
k1 = e
⎛ 36.1± 2.1 kJ / mol ⎞
(17.8± 0.9) −⎜
⎟
RT
⎝
⎠
M −1s −1
k1,298 K = 26M −1s −1
4.3
The observed rate constant data showed good agreement with the expected second order
kinetics. In addition, these data are within the range of typical values for secondary
amines at similar conditions.105 However, the slight upwards curvature of the data
indicates that a multi-step mechanism could also be present. The rate constant may
primarily represent the rate of the limiting step, the HONO dimerization; however, no
literature reports of the kinetics of this reaction were found to confirm this hypothesis.
85
4.4.2 Kinetic evaluation of NaNT 3 formation
The order of the reaction of NaNT 3 formation from DHT 2 and NO2- was determined
by analyzing the results of varying reagent concentrations (Figure 4.6). The decrease of
UV absorbance of HDHT 4 linearly correlated to the increase of absorbance of NaNT 3,
and the measured changes in concentration of nitrite corresponded nearly identically to
initial concentration of 5-AT 1 plus the measured concentration of NaNT 3. No 5-AT 1
was seen in the HPLC traces. Combined with no other peaks being observed in the UV
analysis, this indicates that all of 5-AT 1 was initially reacted to DHT 2 and that only the
desired reactions were occurring in these studies.
Figure 4.6. NaNT 3 reaction order determination through formation of NaNT 3. Diamonds
correspond to initial concentration of nitride of 0.05 M and 5-AT of 0.0167 M; triangles correspond
to the initial concentration of nitride of 0.05 M and 5-AT of 0.033 M; X’s correspond to the initial
concentration of nitride of 0.10 M and 5-AT of 0.033 M.
The initial rate of reaction was approximated by measuring the NaNT 3 concentration
at 25 seconds into the reaction. For the baseline experiment with initial concentrations of
0.0167 M of 5-AT 1 and 0.05 M of NaNO2 (after full conversion to DHT 2, resulting in
0.0167 M of DHT 2 and 0.0333 M of NaNO2, a 1:2 ratio), the initial rate was measured to
be 1.60×10-5 mol L-1 s-1. When the initial concentration of 5-AT 1 was doubled to 0.0333
86
M, keeping the nitrite concentration the same (1:1 ratio of DHT 2 to nitrite), the initial
rate was measured to be 3.20×10-5 mol L-1 s-1, or twice that of the baseline case. When
the initial concentration of nitrite was doubled, keeping 5-AT the same as in the baseline
case (1:4 ratio of DHT 2 to nitrite), the initial rate was measured to be 3.40×10-5 mol L-1
s-1, or slightly over twice that of the baseline case. Combined, these results indicate that,
at the evaluated conditions, the reaction is first-order with respect to DHT 2 and with
respect to HONO, i.e.,
rNaNT = k2 [DHT + ][NO 2− ]
4.4
Equation 4.4 is written in terms of the nitrite ion because, this being a substitution
reaction, only the dissociated nitrite ion participates in the reaction.
Due to the
equilibrium between the nitrous acid (HONO) and the dissociated nitrite ion (NO2-), if the
Sandmeyer type reaction is carried out below pH 3, the concentration of nitrite ion is
insufficient to perform the substitution. Thus, it is necessary to run the substitution
reaction at a higher pH than that used for the diazotization. Unfortunately, under more
basic conditions, the equilibrium between the reactive DHT 2 and the unreactive HDHT 4
favors the non-reactive form. By solving these opposing equilibria (equation 4.5) with
the material balances described by equation 4.6, the product of the concentrations of the
reactive species can be expressed as the product of the initial concentrations of sodium
nitrite and DHT 2 multiplied by the function of pH described by equation 4.7 (the
concentration of HDHT 4 is written as [DHT-OH] for ease of understanding the acidbase equilibrium).
10
⎛ pH − pK
⎞
⎜
⎟
a , NO2− ⎠
⎝
⎡ NO −2 ⎤
⎦ ,
= ⎣
⎣⎡HONO ⎦⎤
10(
pH − pK DHT )
⎡DHT-OH ⎦⎤
=⎣
⎡ DHT + ⎤
⎣
⎦
4.5
−
⎡
⎤
⎡
⎤
⎣ NaNO 2 ⎦ 0 = ⎡⎣HONO ⎤⎦ + ⎣ NO 2 ⎦
⎡ DHT + ⎤ = ⎡ DHT + ⎤ + ⎡⎣DHT-OH ⎤⎦
⎣
⎦0 ⎣
⎦
f ( pH ) =
⎛ pK
⎞
⎜
DHT − pK a , NO − ⎟
2 ⎠
10⎝
⎛ pH − pK
⎞⎤
⎡
⎟
a , NO2− ⎠
⎡1 + 10( pK DHT − pH ) ⎤ ⎢1 + 10⎜⎝
⎥
⎣
⎦⎢
⎣
⎦⎥
87
4.6
4.7
0.04
1.6
[NO2-]
1.4
Relative f(pH)
f(pH)
1.0
[DHT+]
0.8
0.02
0.6
0.4
0.01
pKa
Nitrite
0.2
Concentration (mol/L)
0.03
1.2
pK Diazonium
(estimated)
0.0
0.00
1
2
3
4
5
6
7
8
9
10
pH
Figure 4.7. Reactant concentration dependence on pH at constant temperature and ionic strength; --- represents nitrite concentration, and ....... represents DHT 2 concentration in the baseline
experiment; the solid line represents the combined dependence of the reaction rate on pH scaled to
the maximum rate at constant ionic strength.
The effect of pH on the reactive species concentrations predicts an optimum pH range
in which NaNT 3 can be effectively produced (Figure 4.7). The baseline experiment
concentrations of DHT 2 and NaNO2 were selected.
The pKa of DHT 2 was
approximated as 8, as established in section 4.3.2. However, the reaction variation with
pH, is only valid at a constant ionic strength. Ion interactions and ionic shielding can
play a significant role since this reaction occurs between two ions.
The simplest
approach to modeling ionic strength effects is by the Debye-Hückel limiting law, using
activity coefficients, rather than concentrations, of the reagents.106 This law is limited in
its application, since it is only accurate for binary-ion, highly dilute solutions. However,
more involved models, such as the Meissner corresponding states model107 and Chen
local composition model,108 require knowledge of certain parameters for each ion, many
of which were not found in the literature. Thus, the Debye-Hückel limiting law model
was used to provide at least a working approximation of the reaction dependence on ionic
strength.
88
Because ionic strength is measured on the basis of molality, the volumes of several
reaction mixtures of known concentrations were measured to obtain their densities. It
was found that the reaction mixtures made with different buffered base solutions all had
very similar densities. With the molalities of solutions known, the ionic strength of each
reaction mixture was calculated at the beginning of the reaction (after full conversion of
5-AT 1 to DHT 2 but prior to it reacting to NaNT 3) according to the following equation:
I=
1
2
∑m z
2
i i
4.8
where mi and zi are the molality and charge of the ionic component i.
The dependency on pH and ionic strength is exhibited through modified equilibrium
constants for the dissociations of the two reagents:
γ 1 K a, HONO =
γ 2 K a , DHT + =
[H + ][NO-2 ]
[HONO]
4.9
[H + ][DHT-OH]
4.10
[DHT + ]
where γx is the overall activity coefficient of equilibrium x, calculated as:
log(γ ) =
∑ν
i
log(γ i )
4.11
where νi and γi are the stoichiometric coefficient and special activity coefficient,
respectively, of ion i. The special activity coefficient of each reagent is calculated by the
following equation:109
log(γ i ) = −
zi2 q 2κ
zi2 q 3
=−
8πε r ε 0 k BT
4π (ε r ε 0 k BT )3/2
I
= − Azi2 I
2
4.12
where zi is the charge number of ion species i, q is the elementary charge, κ is the Debye
screening length, εr is the relative permittivity of the solvent, ε0 is the permittivity of free
space, kB is Boltzmann's constant, T is the temperature of the solution, I is the ionic
strength of the solution, and A is a constant that depends on the solvent.
The equilibria in equations 4.9 and 4.10 can now be put in terms of pH and ionic
strength, and, combined with the mass balances in equation 4.6, equation 4.7 instead
takes the form of equation 4.13:
f ( I , pH ) =
(1 + 10
10 pK DHT − pK HONO − 4 A
pH − pK HONO − 2 A I
I
) (1 + 10
pK DHT − pH
)
Thus, the overall reaction rate can be described by equation 4.14.
89
4.13
rNaNT =
(1 + 10
10 pK DHT − pK HONO − 4 A
pH − pK HONO − 2 A I
I
) (1 + 10
pK DHT − pH
)
A r e − Ea / RT [DHT]T [NaNO 2 ]T 4.14
where Ar is the Arrhenius pre-exponential factor, [DHT]T and [NaNO2]T are the total
concentrations of DHT 2 (including HDHT 4) and nitrite (including nitrous acid), and A
is also inversely proportional to T3/2.
0.08
Acetate buffer
Phosphate buffer
1.361
k2 (M-1 s-1)
0.06
1.522
1.471
1.312
0.04
1.832
1.48
1.625
1.857
0.02
1.75
1.44
1.819
2.302
2.6
1.514
1.633
1.548
1.64
1.427
1.104
0
2
3
4
5
6
7
8
pH
Figure 4.8. NaNT 3 generation rate vs. pH and ionic strength. Filled symbols represent
experimental results, and open symbols represent model calculations at corresponding pH and I;
diamonds correspond to buffering with sodium acetate, and triangles correspond to buffering with
sodium phosphate. Data labels indicate the ionic strength at the initial conditions of each
experiment.
To determine A (at room temperature) and KDHT, the pH of the reaction solution was
varied by using different buffered base solutions in the reaction step, as discussed in
section 4.3.2.
Ionic strength was calculated for each of the reaction mixtures.
Additionally, several reactions were run at identical conditions but varying the ionic
strength through addition of NaCl to the buffered base solution. The results are presented
in Figure 4.8. Moreover, for each data point, the predicted reaction rate was calculated
based on equation 4.14, treating the Arrhenius term group as a single constant. These
predicted k values are shown on Figure 4.8 alongside the corresponding experimental
points. Performing a parameter fitting, the following values were obtained:
pK a , DHT + = 7.57 ± 0.07
AR.T . = 0.711 ± 0.026 mol
90
−1
1
2 kg 2
Using the determined values, there is reasonably good agreement between
experimental and calculated reaction rate constant values at pH above 4. Below 4, the
trend is maintained, but the calculated and experimental values diverge – most likely as
the Debye-Hückel model becomes poorer at the more extreme conditions of pH. The
value of the Ka of DHT 2 is in good agreement with the crudely evaluated value by
titration. The value for A is lower than the experimentally obtained value for water of
1.172 mol-1/2 kg1/2;109 but that value represents an ideal, highly dilute solution with small
ionic species.
The temperature dependence of NaNT 3 formation comes both from the Arrhenius
parameters and the effect of temperature on the activity coefficients (and thus, the
dissociation) of the reactive species.
Temperature also affects the pH, but in the
examined range, this effect was deemed negligible.
To account for the effect of
temperature on A, the following equation was considered:
ln k2 = ln ( f ( I , pH ) ) + ln A r −
Ea 1
R T
4.15
Figure 4.9. Arrhenius correlation between temperature and NaNT 3 production rate constant.
91
Because the dependence of f(I, pH) on temperature is known, A was recalculated for
each examined temperature, and the first term of equation 4.15 was subtracted from the
left-hand side to yield a linear plot (Figure 4.9). The Arrhenius rate parameters for NaNT
3 formation were determined to be:
k Arrhen = e
⎛ 25.5± 2.1 kJ / mol ⎞
(11.3± 0.9 ) −⎜
⎟
RT
⎝
⎠
M −1 s −1
4.16
The observed reaction rate constant at room temperature, using a buffer solution
designed to maintain pH between 5 and 4.9 using the baseline experiment concentrations
was 1.9×10-2 mol L-1 s-1. It is interesting to note that the activation energy of the second
step is lower than that of the first step of the direct synthesis, while the reaction rate is
actually lower, as caused by the reaction of two ions occurring in a solution of high ionic
strength (resulting in ionic shielding).
4.4.3 Scale-up to NaNT 3 production
For scaled up direct synthesis of NaNT 3, a set of conditions was chosen based on
information gleaned from the kinetic studies. The nitration of DHT 2 is of first-order in
each of 5-AT 1 and nitrite; thus, the concentrations of both reagents were increased
significantly over those used in the kinetic studies. In addition, NaNO2 was used at a
concentration 4 times that of 5-AT 1 for the following reasons (in addition to requiring 2
equivalents of NaNO2 relative to 5-AT 1 for NaNT 3 synthesis): the diazotization
reaction is second-order in nitrite, and using higher concentrations of it would ensure that
the reaction is complete in a short residence time; 5-AT 1 is a more costly reagent than
NaNO2, making it more desirable to be used as the limiting reagent; and at higher
concentrations, NaNO2 degrades to NOx gas, slightly reducing its available concentration.
Thus, the reagent solutions were prepared for the reaction mixture to have 0.138 M of 5AT 1 and 0.552 M of NaNO2.
The intent of the scale-up was to simulate a process for industrial synthesis. Thus, it
is desirable to produce the target component at the highest possible concentration and in a
solution that can be most easily used in subsequent synthesis steps or out of which it can
be easily separated. Thus, sodium acetate was selected for the buffered base solution
because it was desirable to have a higher concentration of buffered base, leading to
92
higher concentration of reagents in the solution. Additionally, acetate is more processfriendly, both for separating NaNT 3 out of solution and for using the solution directly
downstream for BNCP production.
Conversion of 5-AT to NaNT
1.00
0.95
0.90
0.85
0.80
0.75
0.70
0.65
0.60
0.55
0.50
0
1
2
3
4
5
6
7
Flow rate through zone 2 (mL/min)
Figure 4.10. Correlation of conversion to NaNT 3 with flow rate through a fixed volume; triangles
represent inline quench, and circles represent off-line manual quench.
Figure 4.10 shows the results of the scaled-up flow at room temperature with pH in
the range of 5 to 4 (verified by pH paper as described in section 4.3.2), with both inline
and offline quench. The resultant liquid was amber in color. Gas was evolved in large
quantities, equal to roughly 2 to 3 times the volumetric amount of liquid by visual
observation. Because of this, it was not possible to accurately determine the residence
time of the liquid, as a highly non-linear relationship exists between flow rate and
residence time. Thus, the results are reported in terms of total flow rate through the
nitration zone (5 mL), rather than in terms of residence time.
It can be seen from the figure that, with inline quench, flowing between 580 and 2900
μL/min, there is no significant change in yield of NaNT 3 (non-isolated). Thus, the
reaction is completed quickly, in under 2 minutes, and additional time does not increase,
and may even decrease (possibly due to degradation), the amount of product. Higher
flow rates were not explored due to the limitation of the syringes in terms of pressure. By
removing the inline quench and adding base offline, the pressure drop was reduced
sufficiently to allow the doubling of flow rate to 5800 μL/min. At the highest flow rate,
93
the conversion decreased, which is understandable, given the reduction in residence time.
At 1450 and 2900 μL/min, the conversions were lower with offline quench. This may be
due to lower solubility of NOx at the reduced pressure, causing greater degradation of
NO2- and leading to a slower reaction, or due to pH gradients during the manual addition
of quench, which, if sufficiently high, may degrade both NaNT 3 and DHT 2. No 5-AT 1
was seen in any of the traces. For flow rates at and below 2900 μL/min, no HDHT 4 was
seen in the traces; some HDHT 4 was observed at the highest flow rate, but due to its
instability, no calibration curve was made for it. Because of degradation of nitrite to
NOx, the mass balances using nitrite did not close. However, the method is successful in
safely and efficiently producing NaNT 3 in large yields and at sufficiently high
production rates. Using two syringe pumps and a small bench-top setup, we were able to
successfully produce (in solution) 2.8 grams/hour of NaNT 3 with the best yield of 84%,
and to maximally produce 4.4 grams/hour of NaNT 3 at a yield of 66% from 5-AT 1.
Faster production can be easily achieved given proper equipment, such as HPLC-type
piston pumps capable of high pressure.
With a set of such pumps and reservoirs,
continuous production of NaNT 3 can be easily achieved at much larger flow rates
without greatly increasing the necessary space.
Conversion of 5-AT to NaNT
0.80
0.60
0.40
0.20
0.00
0
10
20
30
Temperature (oC)
Figure 4.11. Correlation with temperature of conversion to NaNT 3 at slightly basic pH.
94
40
The effect of temperature was evaluated by performing the reaction at temperatures
from 5ºC to 35ºC at a flow rate of 2.9 mL/min. Somewhat surprisingly, no significant
differences were seen between the yields. This may be due to the residence times being
affected by changing gas density, compensating for the changes in reaction rate.
However, when the identical reaction was performed using a sub-optimal base solution
(see section 4.3.3 for details) at 0.56 mL/min, there emerged a clear trend (Figure 4.11).
The yield of NaNT 3 was lower, even with longer residence times, but no HDDT 4 or 5AT 1 was seen in the traces. Initially, the reaction solution was at a slightly basic pH. At
increased concentrations, especially at higher temperatures, NaNT 3 and HDHT 4 can
degrade. Thus, the suboptimal conditions may have caused the degradation of HDHT 4,
reducing the available reactive DHT 3, a conclusion that is supported by the lack of
HDHT 4 in the traces and the downward trend of yield with temperature above 21ºC.
Because this reaction is exothermic, performing it in batch by slowly adding base to the
acidic nitrite/DHT 2 solution would result in local pH and temperature gradients, which
may cause degradation of nitrite and of DHT 2, causing suboptimal yields. Thus, there
are clear advantages to performing this reaction continuously with rapid inline mixing.
4.5
Conclusion
Direct synthesis of an energetic compound, NaNT 3, with a highly energetic
intermediate, DHT 2, was achieved using rapid mixing via modular silicon-based laminar
micromixers. Kinetics of the two steps of the reaction were successfully obtained,
including the orders of the reaction and Arrhenius dependence of the first step
(diazotization of 5-AT 1), and the orders of the reaction, dependence on pH, ionic
strength (as a function of temperature), and Arrhenius dependence of the second step
(nitration of DHT 2). The knowledge gained in the kinetic study was applied to design a
scaled-up system to demonstrate production of NaNT 3. At room temperature, with a
small footprint of two syringe pumps plus a small area for two microreactors and tubing,
production of 4.4 g/h of NaNT 3 in solution was performed in a safe manner. It was
demonstrated that by minimizing concentration and temperature gradients, the
performance of the reaction is significantly improved, further confirming the advantages
of continuous flow production of NaNT 3 for both safety and efficacy.
95
Chapter 5.
Epoxide Aminolysis†
Silicon microreactors, in addition to providing a very safe tool for reaction studies,
can provide a wide range of conditions not easily attainable in batch or on the macroscale for reaction evaluation.
However, the reaction knowledge obtained on the
microscale must be transferable to large-scale flow conditions. Therefore, studies are
necessary to determine what effects, if any, result from scaling up reaction conditions
from microreactors.
Epoxide aminolysis was selected as an example chemistry to demonstrate how
microreactors can be used to perform rapid, low-waste condition screening and kinetic
study at high-temperature, high-pressure conditions.
A multi-purpose reactor was
designed to provide thermal isolation of the reactive zone from the inlets, the mixing
region, and the outlets, thus ensuring specified reaction start and end. The robust design
allowed for continuous flow at high pressures, which permitted temperatures far
exceeding the atmospheric boiling points of the used solvents, greatly accelerating the
reaction. The reactor system was used to determine accelerated reaction conditions for a
wide range of epoxide aminolysis chemistries, including critical steps of two actual
pharmaceutical reagents, indacaterol and metroprolol.110, 111 Furthermore, the system was
used to rapidly determine reaction kinetics for a model chemistry, decoupling a side
product synthesis step and allowing for selection of optimized conditions for best reaction
performance. These conditions were then applied at increased flow rates through a
stainless-steel tubular meso-scale reactor, confirming that the reaction proceeds equally
well at larger scales.
†
This chapter describes work done in close collaboration with Matthew W. Bedore, who
at the time was a post-doctoral researcher in the laboratory of Prof. Timothy F. Jamison
in the Department of Chemistry at MIT.
96
5.1
Motivation
The synthesis of β-amino alcohols is an important pursuit in the pharmaceutical
industry and in academic research.
A number of pharmaceutical APIs, including
Oxycontin, Coreg, and Toprol-XL, contain this moiety, while quite a few others, such as
Zyvox and Skelaxin, feature oxazolidones that can be formed through β-amino alcohol
precursors. Frequently, the precursors to β-amino alcohols are difficult to synthesize
and/or very expensive; thus, being able to perform this reaction as rapidly and efficiently
as possible with the highest possible yield is of great interest.
The β-amino alcohol functional group can be assembled by a number of synthetic
pathways, with the one most commonly reported among them being the ring opening of
epoxides with amine nucleophiles.112
While this reaction can, in most cases, be
performed in the absence of a catalyst, it generally proceeds slowly performed at typical
temperatures of solvent reflux.
To attempt acceleration of the reaction, epoxide
aminolysis has been performed in the past by addition of lanthanide triflates,113-116 Lewis
acids,117,
118
solid acid supports,119-122 or by using solvents such as water,123,
124
with
different levels of success and reaction understanding. However, these methods have
been demonstrated primarily on relatively simple substrates, without discussion of the
effects of substrate properties on reaction rates.
The use of elevated temperatures is one of the more general means of β-amino
alcohol formation by the opening of epoxides with amines. Additionally, because of its
inherent simplicity and lack of additional reagents and materials, it remains the preferred
option for industrially practical syntheses due to the ease of application. Elevating the
reaction temperature is a commonly accepted method of reaction acceleration, typically
limited mostly by the stability of the reagents and/or the product. To achieve high
temperatures, either high-boiling solvents are used, or the reaction is pressurized to
increase the boiling point of the solvent. However, solvent properties have a very strong
effect on the reaction rate;125 therefore, it would be highly preferable to select an
appropriate solvent based on its reaction properties and to enable the reaction to be
performed at the highest temperature allowable by the stabilities of the chemical species.
While some work has been done using calorimetry for kinetic studies of epoxide
aminolysis,126-128 few reports have been published analyzing the kinetics and selectivities
97
of this transformation in different solvents or applying kinetic results to process design.129
Additionally, while there have been studies reporting the different kinetic parameters of
primary and secondary amines acting as nucleophiles in the epoxide aminolysis,130 we are
unaware of reports investigating the reactive rate of the β-amino alcohol product itself
reacting with another molecule of epoxide (“bisalkylation”), generally an undesired
process that decreases the yield of the overall desired reaction. Similarly, while it is
conventionally understood that both of the epoxide carbons are reactive in the aminolysis
(thus forming two regioisomers) and that the less sterically hindered carbon will be more
reactive,130-132 few investigations have reported on the ratio of the two products under
different conditions (temperature, solvent effects), which is highly important for
understanding and optimizing an industrial process.
To study a reaction at high temperatures, microwave irradiation is often used, as it is
capable, on the small scale, of rapidly heating the reaction solvent directly. Recently, a
successful demonstration epoxide aminolysis at high temperatures using microwave
heating has been reported,118,
133, 134
demonstrating the applicability of elevated
temperatures and pressures to this synthesis. Microwave chemistry has been used to
achieve such conditions for rapid reaction monitoring;135 however,
microwave
irradiation is limited in its penetration depth, making scale-up to industrial levels highly
difficult.
Additionally, due to the limitation of microwave-transparent materials,
microwave chemistry vessels are typically limited regarding pressurization capability.
There have been reports of continuous-flow microwave processes, in attempts to
overcome scaling issues.136-139
However, these often require complicated and/or
unwieldy microwave generators.
Properly designed microreactors can often reach or surpass the pressure, temperature,
and heat transfer capabilities obtained in traditional microwave processes.140,
141
Continuous-flow microreactors also eliminate the need to scale up hazardous batch
reactions, provide chemical synthesis understanding at conditions replicable at large
scales, and are simpler to operate than microwave reaction systems.15 Microreactors for
continuous-flow syntheses are increasingly more applied in both academia and the
pharmaceutical industry,142-145 often used to more efficiently produce biologically active
materials.146 As compared to conventional batch processes, microsystems enable rapid
98
heat and mass transfer, resulting in improved reaction profiles for more accurate kinetic
studies (see Chapter 4). In addition to safely enabling high temperatures and pressures
not easily attainable in batch chemistry while allowing for rapid reaction monitoring,143
microreactors are highly efficient in the use of reagents for kinetic screening studies.15, 19
5.2
Epoxide Aminolysis Synthesis
The formation of β-amino alcohols by epoxide aminolysis (see Scheme 5.1 for the
general reaction schematic) is currently a very pharmaceutically prevalent reaction,
typically performed as a batch process, although it has been performed in a continuous
manner in one instance.147 However, our investigation is, to the best of our knowledge,
the first study of epoxide aminolysis completed in a microfluidic system.
R3
O
+
R1
HN
R2
R3
R3
N
Δ
N
R2
R2
+
R1
OH
P
R1
OH
R
Scheme 5.1. General epoxide aminolysis synthesis, with two possible products (designated with P for
the desired product and R for the regioisomer).
As can be seen in Scheme 5.1, there are two products in an epoxide aminolysis
reaction, leading to certain quantities of an undesired side-product.
The selectivity
towards the desired product depends on several factors, with the most important being the
steric hindrance afforded by the R1 group in the scheme, although that factor is an
intrinsic property of the selected reagent and is not controllable in the process. However,
other conditions (temperature, solvent, co-solvents) also play a role, which is of great
interest because an understanding of these effects can afford knowledge towards better
industrial process design.
If the amine nucleophile selected for the synthesis is a primary amine, then the
product, which is thus a secondary amine, can also react with the epoxide in a similar
reaction (see Scheme 5.2). Therefore, in addition to the formation undesired regioisomer
side-product, the reaction yield will be further reduced by the product participating in an
99
undesired reaction. Thus, as in the case of the studied synthesis reaction of styrene oxide
5 (SO) and 2-aminoindan 6 (AI), in addition to the desired product 7, up to four other
side-products (8-11) can also be present.
Scheme 5.2. Expanded epoxide aminolysis reaction scheme when a primary amine is used as the
nucleophile. Styrene oxide 5 and 2-aminoindan 6 are shown as the example reagents; β-amino
alcohol 7 is the desired product.
One of the reasons for the strong interest in β-amino alcohol synthesis studies is its
importance to pharmaceutical active ingredients such as indacaterol 12 (Scheme 5.3), a
novel β-adrenoceptor agonist developed by Novartis.148 This drug candidate is used in
the treatment of chronic obstruction pulmonary disease (COPD) and has shown a great
deal promise as a one-dose-daily inhaled bronchodilator,149 making it potentially more
patient- friendly than currently existing several-doses-daily treatments. Indacaterol is
currently approved for use in the European Union and is undergoing approval review by
the FDA. Being an inhaled API, its relative composition in a dose is extremely small,
making the relative volume of production of this compound rather low. Thus, it is
particularly well-suited to continuous-flow production using micro- or meso-scale flow
systems.
The reported current synthesis of 12 centers on the aminolysis of epoxide 13 with
amine 14 to afford precursor 15 under a protracted reaction time of 15 h.150 In addition,
the regioisomer 16 and a product of double alkylation 17 are also formed in significant
100
quantities. Thus, the currently reported reaction time is extremely slow for continuousflow applications, leading to interest in significant reaction acceleration and study using
microfluidic techniques.
Scheme 5.3. Synthesis of 15 as a precursor to indacaterol 12, showing conditions and product
distributions reported in the patent to indacaterol 12. 150
Another useful β-amino alcohol API is metoprolol, used in the treatment of
hypertension151 and licensed under a variety of different names, including Lopressor
(Novartis AG) and Toprol XL (AstraZeneca).
The synthesis centers around the
aminolysis of the readily available epoxide 18 with isopropylamine 19 to form
metoprolol 20 (Scheme 5.4).
The epoxide aminolysis is typically performed using
multiple equivalents of isopropylamine at reflux in a polar protic solvent, with reaction
times ranging from 2 to 5 h, and with 65-70% yield of desired product.152-155 Similarly to
indacaterol precursor 15, it is highly desirable to affect reaction acceleration without
sacrificing product yield.
There have been numerous reports of attempts to accelerate epoxide aminolysis.
Attempts to catalyze the aminolysis with solid acid supports such as PMA-alumina,122
Amberlyst-15,121 and ZnClO4-alumina119 led to little or no product formation even after
extended periods of time. Catalysis with lanthanide triflates such as Er(OTf)3115 and
Yb(OTf)3114 ultimately led to shorter reaction times (approx. 5 h) but yielded large
amounts of undesired byproducts.
Thus, this study focuses on application of high
101
temperatures at elevated pressures, either by direct application of heat in a silicon
microreactor or in a microwave vial to provide a comparison to batch synthesis.
Scheme 5.4. Synthesis of metoprolol 20 from epoxide 18 and isopropylamine 19.
5.3
Microchemical System Design
The primary functionality of the microreactor in this synthesis is the capability to
achieve high temperatures (up to 250ºC) at elevated pressures (~35 bar). In batch, high
pressures are typically achievable in sealed metal vessels; however, sampling at these
high pressures is often a challenge, and each set of reaction conditions or compositions
requires preparation of a separate batch. In contrast, sampling from continuous-flow
microsystems is extremely simple, as the liquid-phase flow reaches ambient pressure
immediately after passing through the system pressurization device. Additionally, by
simply modifying the flow rates of each component or solvent or by changing the
temperature, a large number of experiments can be performed with a single set of
prepared reagent solutions.
5.3.1 Microreactor Design
The reactor used in this study (Figure 5.1) was fabricated in silicon due to its high
thermal conductivity, enabling rapid thermal equilibration and precise temperature
control.2 Moreover, the rigidity of silicon easily allows for the 35-bar pressure applied in
this study to access the desired temperatures.
When accessing the desired high
temperatures, it must be ensured that the fluidic connections maintain the seal, especially
considering the high pressures applied in this study. Thus, it was decided to thermally
separate the portion of the chip containing the inlets and outlets from the main reaction
zone (see Figure 5.1a), based on a configuration previously applied to synthesis of
quantum dots at supercritical fluid conditions.156 This enabled compression packaging to
102
be used (see sections 2.2.1 and 3.3.3), cooling the compression chuck to ensure that the
polymer o-rings retain their integrity. An additional benefit of this configuration is the
ability to mix the reagents well using a mixing zone before they are heated (thus, before
the reaction begins). The active reaction volume of the reactor was 120 µL, in addition to
20 μL of the mixing zone.
(a)
Inlets
Outlet
(b)
Cooled
section
Mixing
zone
Heated
section
Reaction
zone
cm
Figure 5.1. Two-thermal-zone microreactor: (a) illustration, (b) photograph.
Because epoxide aminolysis is a simple coupling of two reagents with no catalyst or
specific pH, the easiest way of stopping the reaction is by reducing the temperature of the
reaction stream. On the microscale, this happens rapidly (see the next section) as the
reaction flow transitions from the heated zone to the cooled one. Combined with the premixing, the isolation of the heated zone from the cooled one ensures highly accurate
knowledge of reaction residence time given a known flow rate. Thus, the advantages
afforded by the microscale silicon reactors enable accurate kinetics determination.
The silicon channels were coated with silicon nitride to provide chemical resistance,
enabling the reactor to withstand slightly basic conditions at high temperatures, as well as
all acids and organic solvents, thus providing a chemically and physically robust
environment. Although the use of silicon nitride is a slight departure from typical glass
systems, the significantly higher resistance of nitride to caustic corrosion, as compared to
that of oxide, is a major advantage, expanding the utility of the microreactor to a wider
103
range of chemical systems. Additionally, as the microfabrication process of nitride
deposition is no more difficult than that of silicon oxide growth, there is no inherent
drawback to its usage over that of oxide.
The
microreactor
was
fabricated
using
standard
silicon
micromachining
techniques,157 in a process nearly identical to that of the 2nd-generation micromixers
(section 3.3.2). Channel layout was defined by photolithography and realized by DRIE in
a silicon wafer (15-cm diameter; 0.65-mm thickness) to a depth of 400 μm. A silicon
nitride layer (500 nm) was grown on the silicon surface, and the entire device was capped
and sealed by anodically bonding a Pyrex wafer (1.0-mm thickness).
5.3.2 Continuous-Flow System Setup
To enable the fluidic connections to the microreactor, the section of the reactor
containing the inlets and outlet was compressed in a custom microfluidic chuck machined
out of aluminum. Combined with Kalrez® (a perfluoroelastomer) o-rings (Z1028 FFKM,
size 005, Marco Rubber) used to seal the fluidic connections, the reactor system is thus
capable of withstanding a wide range of solvents and chemical conditions (see section
3.3.3 for discussion). The elasticity of the o-rings enabled fluidic sealing by compression
to pressures of over 100 bar.158
The fluidic compression chuck had two channels 3/16” in diameter drilled through its
length (along the width of the reactor) for cooling via house cooling water. The chuck
was machined with 10-32 ports coned-bottom ports, and PEEK fittings were used
(Upchurch NanoTight® headless fittings, F-333N), connecting to 1/16” OD, 0.020” ID
PEEK tubing. For experiments requiring only two inlet flows, the third inlet, which
remained unused, was capped with a PEEK plug (Upchurch P-550). Inlet tubing was
connected to 8-mL high-pressure stainless steel syringes (702267, Harvard Apparatus),
which were independently driven by two to three syringe pumps (PHD 2200 or PHD
Ultra, Harvard Apparatus). Thus, reaction time was controlled by modifying the total
flow rate entering the reactor (consisting of the sum of the flow rates of the pumps). The
outlet tubing was connected to a backpressure regulator, either 250 psi (U-608,
Upchurch) or 500 psi (U-609, Upchurch).
104
Figure 5.2. Photograph of assembled microreactor system, showing the Si microreactor packaged in
compression and heating chucks, with coolant and reagent flow lines connected and the heating
system installed.
The reaction zone of the reactor was compressed between a 3/8” thick piece of
borosilicate glass and a 1/16” thick piece of graphite, which was in direct contact with a
custom-machined aluminum heating chuck. The heating chuck was drilled with two
holes for insertion of 1/8”-diameter cartridge heaters (35 W, 120 V, CSS-01235/120V,
Omega) and a 1-mm-diameter hole for a wire thermocouple (K-type, SC-GG-K-30-36,
Omega), placed 0.5 mm beneath the chuck surface. The thermocouple provided data to a
PID controller (CN742, Omega), which controlled the cartridge heaters via a solid-state
relay (SSRL240DC10, Omega). The system is shown in Figure 5.2.
5.3.3 Heat Transfer Analysis
The high thermal conductivity of silicon (148 W/m·K compared to stainless steel ~40
W/m·K) greatly aids in spreading heat and significantly reduces the occurrence of hot
spots. The use of aluminum for the heating chuck and of graphite as the liner between
105
the chuck and the reactor further helped distribute the heat while providing high heat
transfer. A thermocouple was inserted into the heating chuck 0.5 mm below the chuck
surface. To confirm the temperature distribution, finite element modeling was performed
(Comsol Multiphysics 3.2). A 2-D lengthwise cross-section of the reactor was simulated,
including the heating and compression chucks. A temperature of 455 K (182ºC) was
assumed for the cartridge heaters, and a temperature of 285 K (12ºC) was given for the
boundary between the compression piece and cooling fluid. The overall results of the
simulation are given in Figure 5.3.
Figure 5.3. Finite element modeling of heat transfer in the reactor setup.
It can readily be seen that the etched-out area of the reactor establishes thermal
separation between the inlet/outlet area (including the mixing zone) and the reaction area
of the reactor. This allows the area in contact with polymer o-rings and fittings to remain
at room temperature when the compression chuck is water-cooled, while the reaction
zone is at temperatures of up to 300°C. Additionally, the temperature at the location of
the thermocouple is within 0.02ºC of that of the cartridge heaters.
Figure 5.4 shows the cross-sectional temperature profile along the reactor at the depth
half-way into the channels. Here, the distance of 0 cm represents the point at the haloetched gap, and 4.0 cm represents the bottom of the reactor. It can be seen that, at a
106
maximum temperature of 182ºC, there is a relatively small variation of 0.5ºC across the
length of the reactor.
Additionally, the spiral channel layout ensures that any
inhomogeneity in temperature would have little effect on the reaction, as fluid traverses
the length of the reactor multiple times in the course of one residence period.
455
Temperature (K)
454.9
454.8
454.7
454.6
454.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
Reactor Position (cm)
Figure 5.4. Cross-section of the temperature profile along the reactor at the channel depth.
In addition to enabling simple fluidic packaging, the small volumes within the
microreactor ensure that the reaction mixture is rapidly brought to room temperature
upon leaving the reaction zone, providing highly efficient quenching and accurate
residence time evaluation.
This can be confirmed by performing heat transfer
calculations. There have been a number of experimental investigations of heat transfer
into and out of fluid through silicon microchannels, as reviewed by Morini.159 Many
different approaches and correlations for heat transfer have been developed, with a great
deal of discrepancy. A recent investigation by Park and Punch has performed theoretical
derivations, computational simulation, and experimental evaluations to derive a
correlation between the Nusselt number and the various properties of the flowing fluid:160
Nu = 0.015Br −0.22 Re0.62 Pr 0.33
5.1
where Nu is the Nusselt number, Br is the Brinkman number, Re is the Reynolds number,
and Pr is the Prandtl number, respectively defined as follows:161
107
Nu =
Br =
hDh
k
μU 2
k (Tw − T0 )
Re =
UDh ρ
Pr =
μ
Cˆ p μ
k
5.2
5.3
5.4
5.5
where h is the heat transfer coefficient in the system, Dh is the hydraulic diameter, k is the
thermal conductivity of the fluid, μ is the fluid viscosity, U is the linear velocity of the
flow, Tw and T0 are the wall and fluid bulk temperatures, respectively, ρ is the fluid
density, and
Cˆ p
is its mass heat capacity. The hydraulic diameter Dh is a function of the
channel width w and depth d as follows:
Dh =
wd
2( w + d )
5.6
For the microreactor system, taking ethanol as the fluid, and considering the channels
of 400 × 400 μm, the Nusselt number can be calculated (properties of ethanol at room
temperature were used; although the properties will change somewhat with temperature,
the overall effect on the value of h will not be significant). For the target temperature of
180ºC, with fluid flows from 4 μL/min to 360 μL/min (residence times of 30 minutes to
20 seconds), the resulting Nusselt number ranges from 0.53 to 1.25, respectively.
According to work by Wu and Cheng,162 the Nusselt number would also be affected
by channel surface roughness and the hydrophilicity of the surface. However, they found
that at laminar flow rates, the Nusselt number with a silicon oxide surface is only ~1.5%
lower than with a bare silicon surface (silicon nitride is more similar to silicon oxide than
to silicon). Additionally, they found that surface roughness plays a role in heat transfer,
as well. For a surface roughness of 1 μm in a 400-μm channel (see section 2.1.1), this
would decrease the Nusselt number by ~18%. Thus, the Nusselt numbers found based on
the correlation by Park and Punch160 should be adjusted downward by 20% for the
slightly rough (due to DRIE) silicon-nitride-covered channels.
108
Because the Nusselt number is of the same order of magnitude for flows varying by
two orders of magnitude, heat transfer effects will be most significant for fastest flow
rates. Thus, for 360 μL/min of flow, the Nusselt number is taken to be 1 (based on the
calculated value and the above correction), which produces a heat transfer coefficient of
1690 W/m2·K. The time required for the fluid to equilibrate with the walls can be
obtained from the following equation:
hAs
t
−
T − Tw
ˆ
= e ρVCP
Tinitial − Tw
5.7
where As is the surface area in contact (taken as the three silicon walls of the channel), V
is the volume being equilibrated, and Tinitial and T being the temperature of the fluid
initially and at time t, respectively. Thus, the temperature of the fluid is within 1ºC of the
wall temperature within 0.09 s, or 0.45% of the residence time. Therefore, we can
confidently state that thermal equilibration of the fluid is very rapid.
5.4
Reaction Screening and Profiling
5.4.1 Experimental Setup and Operation
General Methods
All chemicals, unless specified otherwise, were reagent-grade and used as received.
AI 6 was purchased from TCI America (Portland, OR). All other purchased reagents
were obtained from Sigma-Aldrich (St. Louis, MO). The indacaterol substrates 13148, 150,
163-165
and 14166 were prepared according to literature procedures. The epoxide 18 for
metoprolol was synthesized from the corresponding phenol and epichlorohydrin
according to published reports.153
For initial reaction screening, all aminolysis reactions were initially performed as 1 M
solutions in ethanol using a Biotage Initiator single cavity microwave reactor under
normal absorption and in 0.5-2 mL sealed vials (5 mL total volume). The products were
then separated either with preparative TLC on precoated silica gel 60 F254 glass sheets or
by column chromatography on Silicycle silica gel (230-400 mesh), eluting with
hexane/ethyl acetate or dichloromethane/methanol.
109
A microwave reactor was selected for the initial testing because it allows for some
limited pressurization of the vessel, heating organic solvents to relatively high
temperatures. Moreover, the heating profile in a microfluidic reactor is more similar to
that in microwave reactors than to that observed in typical batch heating, considering the
time to reach desired reaction temperature. However, it should be noted that the cooling
profile in microfluidic reactors is far superior to that generally observed in batch
microwave reactors. Additionally, the microreactor setup is capable of operating at
significantly higher temperatures than those achievable in the microwave due to higher
pressure tolerances.
Analytical
All components were analyzed by 1H and
13
C NMR spectroscopy using a Bruker-
Avance 400 MHz spectrometer and compared to known literature compounds when
available. HPLC quantitative analysis was performed on an Agilent 1200 Series LC/MS
using either an Eclipse XDB-C18 or a Zorbax Eclipse Plus C18 reverse phase column, a
methanol/water mobile phase, and a 254 or 210 nm wavelength detector. Yields were
calculated based on normalization of response factors using naphthalene as an internal
standard. GC quantitative analysis was performed on an Agilent 7890A GC system.
Yields were calculated based on normalization of response factors using dodecane as an
internal standard.
Microreactor Epoxide Aminolysis Procedure
The system described in section 5.3.2 was used for this procedure. A solution of the
desired epoxides (10 mmol) and napthalene (internal standard, 10-20 mol%) was diluted
to 5 mL with ethanol and placed in an 8-mL high-pressure stainless steel syringe. A
solution of the amine (12 mmol for 1.2 equivalents) was diluted to 5 mL with ethanol and
placed in a separate 8-mL syringe before being connected to the microreactor. Ethanol
was chosen as the initial solvent due to its good solvating properties, high dielectric
constant, and low toxicity.
For the formation of the indacaterol precursor 15, the epoxide 13 (234.2 mg, 0.79
mmol) and naphthalene (internal standard, 15.7 mg, 0.120 mmol) were dissolved in N-
110
methylpyrrolidone (NMP) (1.8 mL), and the suspension was gently heated to dissolve the
solid. After cooling, H2O (200 µL) and amine 14 (181.7 mg, 0.96 mmol) were added to
the mixture and stirred before being placed into a single 8-mL high-pressure stainlesssteel syringe and connected to the microreactor.
The reagent stream(s) was/were pumped through the microreactor, pressurized by a
250- or 500-psi backpressure regulator, at identical flow rates, and reaction times and
temperatures were varied.
Following every change in reaction conditions, five
microreactor volumes (5 × 120 µL) were allowed to pass through the outlet to achieve
steady state and flush through the post-reactor section before samples were taken for
quantitative analysis.
5.4.2 Results and Discussion
Fluid superheating
When applying heat to the microreactor, the onset of boiling was observed to occur at
significantly higher temperatures than predicted by vapor pressure calculations. Using
the Antoine equation, the boiling points for ethanol and acetonitrile were calculated at
250 psi to be at 174°C and 208°C, respectively, and at 500 psi, 206°C and 259ºC,
respectively. However, within the microreactor, pure ethanol was not observed to boil at
250 psi until 217°C was attained, with freshly incoming material ceasing to boil when the
reactor was cooled to 206°C. At 500 psi, ethanol did not boil until 250°C, with flashing
ceasing when cooled to 246°C. A similar effect was observed with acetonitrile, which, at
250 psi, only boiled at 246°C, ceasing at 239°C. At 500 psi, boiling was not achieved
even when heated to 300°C. Because interfacial forces are dominant at the microfluidic
scale and combined with the smoothness of the channel walls, solvent superheating to
above boiling temperatures was obtained. Thus, the dominance of interfacial forces on
the microscale further extends the range of operating temperatures beyond even those
afforded by the pressurization.
Boiling was easily observed visually, as the top side of the microreactor consists of
transparent borosilicate glass, allowing an unfettered view into the reaction channel and
further demonstrating the utility of the applied reactor design. When it occurred, boiling
was observed at the transition between the cooled mixing zone and the heated reaction
111
zone, where the reaction stream flashed, or was rapidly transformed primarily into gas
phase with only small volumes of liquid phase remaining.
When temperature was
sufficiently decreased, flashing was seen to cease, with the reaction stream remaining
homogenous (liquid-phase) throughout the reaction zone.
5.4.3 Model Chemistries
To test the effectiveness of microreactors in the aminolysis of epoxides, as well as to
observe how different moieties and conditions affect the reaction, we applied the system
to a variety of substrates, both epoxides and amines (shown in Scheme 5.5). We also
wanted to directly compare the results obtained under standard batch microwave
protocols with those obtained using the microreactor at corresponding conditions to
provide a direct flow-to-batch comparison. The results are summarized in Table 1.
Scheme 5.5. Epoxides and amines used as model substrates.
The aminolysis of phenyl glycidyl ether 22 with 2-aminoindan 6 under microwave
irradiation in a sealed vial went to completion in 30 min at 150°C (Table 5.1, Entry 1).
The pressure in the 5-mL vial with 1 mL of solution ranged from 100 to 130 psi over the
course of the reaction. Only minor isolated amounts (~1-2%, not quantified by HPLC
analysis) of the regioisomer were observed in this reaction. Formation of the bisalkylated
side product due to the subsequent reaction of the main product with the epoxide was
observed, with excellent overall mass balances.
112
Table 5.1. Epoxide aminolyses performed in a microreactor and compared to microwave (μw) batch.
Epoxide
Amine
NH2
O
OPh
22
6
O
NH2
OPh
22
Entry
Conditionsa
(psi)
Amine
equiv
Temp
(oC)
Flow rateb
( L/min)
Time
Productc
( -Opened)
(%)
Isomer
( -Opened)
(%)
Bisalkyld
(%)
conv.
(%)
1
Batch ( w)e
1.2
150
-
30 min
72
-f
26
> 99
2
reactor (250)
1.2
150
4
30 min
73
-
26
>99
3
reactor (250)
1.2
195
60
2 min
72
-
24
98
4
reactor (250)
1.2
195
120
1 min
71
-
21
93
5
Batch ( w)e
1.2
150
-
30 min
72
-
25
> 99
6
Batch ( w)g
1.2
150
4
30 min
74
-
24
98
7
reactor (250)
1.2
195
12
10 min
70
-
22
93
8
reactor (250)
1.2
195
24
5 min
63
-
18
82
9
Batch ( w)e
1.2
150
-
30 min
75
-
24
>99
10
Batch ( w)g
1.2
150
-
30 min
82
-
17
>99
11
reactor (250)
1.2
150
4
30 min
82
-
16
>99
12
reactor (250)
1.2
195
40
3 min
82
-
13
98
13
reactor (250)
2.0
195
120
1 min
84
-
6
92
14
Batch ( w)e
1.2
150
-
30 min
57
7
21
90
15
Batch ( w)e
1.2
150
-
30 min
62
10
19
97
16
reactor (250)
1.2
150
4
30 min
62
7
16
94
17
reactor (250)
1.2
195
24
5 min
60
8
14
91
18
reactor (250)
2.0
195
24
5 min
68
9
8
91
19
Batch ( w)e
1.2
150
-
30 min
54
-
-
58
20
reactor (250)
1.2
150
4
30 min
39
-
-
40
21
reactor (250)
1.2
195
4
30 min
66
-
-
72
22
reactor (500)
1.2
245
4
30 min
71
-
-
93
23
Batch ( w)e
5.0
150
-
30 min
19
3
-
22
24
reactor (250)
5.0
150
4
30 min
15
2
-
17
25
reactor (500)
5.0
240
4
30 min
68
6
-
78
23
O
NH2
OPh
22
O
24
NH2
Ph
5
24
O
NH
25
O
Ph
27
26
NH2
28
a
All reactions were run in ethanol at 1 M concentration in epoxide. bCombined flow rate of both reagents. cAll yields are calculated by HPLC analysis with an
internal standard with the exception of Trials 10-14 which were analyzed by GC. dBis-alkylation arises from product reaction with starting material to give the tertiary
amine. e1 mL volume in a 5 mL vial. f ~1-2% of regioisomer was isolated but not quantified. g2 mL volume in a 5 mL vial.
113
Using the flow microsystem with 250-psi backpressure and a flow rate of 4 µL/min
(30-min residence time) at 150°C, complete conversion was also obtained, and product
distribution mirrored that of the microwave experiment (Table 5.1, Entry 2). At this
pressure, ethanol was easily heated to 195°C without boiling being observed in the
microreactor, and near-complete conversion was realized at this temperature in 2 min
(Table 5.1, Entry 4), demonstrating significant reaction acceleration.
Additionally,
Figure 5.5 shows that the amount of excess of amine has a significant effect on product
distribution, with greater amine concentration leading to improved yield and selectivity.
Similar results for flow-to-batch comparison were observed with phenyl glycidyl ether 22
and aniline 23 (Table 5.1, Entries 5-8), although this reaction has a smaller rate increase
with temperature.
100
Conversion (%)
80
60
Product
Bisalkylation
Total
40
20
0
0
1
2
3
4
Residence Time (min)
5
Figure 5.5. Aminolysis of 22 with 6 in microreactor at 195ºC and 250 psi; open and closed symbols
represent 1.2 and 2.0 initial molar equivalents of amine 6, respectively.
When performing this reaction in batch using the volatile tert-butylamine 24 (boiling
point of 46°C), the product distribution appeared to be dependent on the fill volume of
the vial (Table 5.1, Entries 9-10, 14-15). Its reaction with phenyl glycidyl ether 22 was
complete after microwave irradiation for 30 min. However, an increase in the amount of
bisalkylation product was observed when 1 mL of solution was heated in a 5-mL sealed
vial as compared with 2 mL of solution. A similar effect was seen in aminolysis of
114
styrene oxide 5 with tert-butylamine 24. This variance is likely due to the difference in
available headspace, which leads to variation in the amount of amine in the vapor phase,
with more amine having evaporated from solution with greater headspace. Because the
amine is more volatile than the solvent, its vaporization decreases its concentration in
solution, reducing the reaction efficiency.
In contrast, the absence of headspace in the continuous-flow microreactor led to
consistent product distributions (Table 5.1, Entries 11 and 16).
Performing these
reactions at 195°C (enabled by the 250-psi backpressure) resulted in almost complete
conversion of phenyl glycidyl ether 22 at a residence time of 3 min (Table 5.1, Entry 12)
and a conversion of 91% SO 5 at a residence time of 5 min (Table 5.1, Entry 17).
Internal and trisubstituted epoxides were also examined under microreactor
conditions. Using 1,4-dihydronaphthalene oxide 25 and the hindered secondary amine
indoline 26, aminolysis was conducted both in the microwave and microreactor at 150ºC
(Table 5.1, Entries 19 and 20). Only moderate substrate conversion was obtained in each
case, with higher conversions observed in the microwave process compared to the
microreactor, which can be attributed to two factors. First, because the solvent in this
reaction is more volatile than the substrates, the overall concentration in a microwave vial
is somewhat higher than nominal due to the headspace available to volatilize the solvent.
Second, microwave reaction times are slightly extended above the nominal reaction time
due to periods of warming and cooling during the pre- and post-reaction phases. Using a
500-psi backpressure regulator in the microreactor setup allowed for heating of ethanol to
245ºC while maintaining liquid phase. At this reaction temperature, nearly complete
conversion was observed in 30 min; however, the appearance of a new unidentified
byproduct was observed by HPLC analysis. It is probable that degradation of the product
occurs at such high temperatures. It is notable that temperatures approaching 245°C for
ethanol are not attainable in microwave batch reactions due to the pressure limitations of
the microwave system.
Ring opening of trisubstituted epoxides has also been a challenge in microwaveassisted aminolysis of epoxides.118 For the aminolysis of 1-phenylcyclohexene oxide 27
with propylamine 28, the microreactor, predictably, gave results similar to those in batch
at comparable conditions, with poor conversions, even with a large excess of the amine
115
(Table 5.1, Entries 23 and 24). However, the use of the 500-psi backpressure regulator
enabled a reaction temperature of 240ºC, affording moderate conversions after a 30-min
residence time (Table 5.1, Entry 25). The use of 5 equiv. of amine represents almost a
neat amine solution in one syringe; however, high reaction temperatures could still be
maintained in the microreactor without flashing.
Scheme 5.6. Aminolysis of SO 5 with aniline 23; product 30 is often favored.
31
30
29
Figure 5.6. Product distributions of aminolysis of SO 5 with 23 at different reaction conditions.
The opening of SO 5 with aniline 23 (Scheme 5.6) is a unique example because
selectivity for the terminal over the benzylic position can be poor.123, 124 Indeed, a batch
microwave reaction in methanol led to aminolysis favoring the attack on the benzylic
position (giving 30) over the terminal position (leading to 29) (Figure 5.6). Using
solvents such as ethanol and isopropanol resulted in a reaction favoring the terminal end
of the epoxides due to the hydrogen bonding properties of these solvents affecting the
116
electronic coordination. Interestingly, increasing the temperature in the microreactor
from 150ºC to 245ºC also gave a reversal in selectivity, with attack favored at the
terminal end of the epoxide. This further demonstrates the utility of microfluidic systems
for easily studying reactions at conditions otherwise not readily attainable.
To study the effect of solvent on the aminolysis of a system such as indacaterol
(Scheme 5.3), we selected as a model system the aminolysis of SO 5 with AI 6 to attain a
similar electronic and steric environment as the reaction between 2 and 3. Heating of this
reaction mixture in the microwave at 150ºC for 30 min led to complete conversion,
giving 59% of the desired product 22 and significant amounts of the regioisomer 23 and
bisalkylation side products 24 and 25 (Scheme 5.2). The overall selectivity for terminal
over benzylic reaction of the epoxide was similar to, although slightly lower than, the
indacaterol reaction in diglyme.
Product Yield (%)
80
60
EtOH, 195ºC
MeCN
MeCN/EtOH 9:1
MeCN/EtOH 3:1
MeCN/MeOH 9:1
MeCN/H2O, 9:1
40
20
0
0
5
10
15
20
25
Residence Time (min)
30
Figure 5.7. Solvent study of SO 5 aminolysis with 1.2 equiv. of AI 6, at 240ºC, except in ethanol
(performed at 195ºC).
It has been reported that polar aprotic solvents can improve selectivity in aminolysis
reactions at the expense of overall reaction rate, as compared to polar protic solvents such
as ethanol.118, 167 The study of solvent selection was aided by two particular advantages
of microreactor flow technology. First, by altering temperature and flow rate, reaction
117
conditions can be scanned quickly to find optimum conversion and product yield.
Second, due to the absence of headspace, mixtures of polar protic and polar aprotic
solvents can easily be employed without concern for the relative boiling point of each
component. In this manner, we could consider a polar protic solvent as a potential
promoter for the reaction occurring primarily in a polar aprotic solvent.
As reported in literature,125 the reaction rate depends on the solvent properties of
polarity, polarizability, and, to much lesser extents, electrophilicity and nucleophilicity.
Our results of a solvent screen fit well with the reported trends. Figure 5.7 shows the
results of the microreactor-enabled solvent screen of SO 5 aminolysis with AI 6, using
1.2 equiv. of the amine. Using ethanol as a baseline, the microreactor aminolysis was
nearly complete in 5 min at 195ºC to afford 59% of 7 along with 14% of the regioisomer
8. Switching to acetonitrile as the solvent and operating with a 250-psi backpressure
regulator, temperatures up to 240ºC were obtained before flashing of the solution was
observed in the microreactor. Even at this increased temperature, product yields were
considerably lower when compared to those in ethanol at similar residence times.
However, a 30-min residence time resulted in completion of the aminolysis, and up to
69% of 7 was obtained, as quantified by HPLC analysis. The increase in overall product
yield was derived mainly from the improved regioselectivity of the reaction, as attack at
the terminal position of the epoxide over the benzylic position is favored. Incorporation
of a 9:1 mixture of acetonitrile to ethanol in the microreactor efficiently accelerated the
reaction to where conversions of 99% were achieved in only 15 min. Yields of 7 were
maintained at a high level (68%).
Changing the solvent system to either 75:25
acetonitrile/ethanol or 9:1 acetonitrile/methanol also gave improved conversions at
comparable residence times. Finally, using a ratio of 9:1 acetonitrile/water, conversions
at similar time intervals surpassed those obtained in pure ethanol at 195ºC, and nearly
complete aminolysis was observed at a 10-min residence time with 66% yield of 7.
5.4.4 Application to Pharmaceutical Compounds
Application to metoprolol.
Following our investigation of model epoxide aminolysis reactions using a
continuous-flow microreactor, we intended to highlight this approach through the
118
formation of pharmaceutically relevant β-amino alcohols. We first chose metoprolol 20,
as discussed in section 5.2, due to its rather simple structure to illustrate the efficiency of
epoxide aminolysis using the developed method.
The epoxide aminolysis of metoprolol 20 is typically performed using multiple
equivalents of isopropylamine at reflux in a polar protic solvent, with reaction times
ranging from 2 to 5 h.152-155 In examining batch microwave conditions, we again noted
that the amount of 20 and bisalkylation side product 21 were dependent on reactor
headspace due to the low boiling point of isopropylamine. A typical product yield was
65-69%, with complete conversion in 30 min at 150ºC. Under microreactor conditions,
loss of the volatile amine at high temperatures was not a concern, as discussed
previously; accordingly, we focused on maximizing conversion and throughput.
At 500 psi, temperatures up to 240ºC were achieved before flashing of ethanol was
observed in the microreactor.
As shown in Figure 5.8, increasing the amount of
isopropylamine at this temperature led to decreases in both bisalkylation and the reaction
time, with complete reaction occurring in as short as 15 s, with 91% yield of the desired
product.
Under these conditions, a single 120-µL microreactor working under
continuous-flow operation is capable of delivering 7.0 g/h (61 kg/year) of metoprolol.
Operating 17 microreactors in parallel or scaling to a only a 2-mL continuous-flow
reactor could ultimately produce over 1 metric ton of this important drug per year.
100
90
80
2.0 Amine equiv.
4.0 Amine equiv.
1.2 Amine equiv.
70
Ratio of Product Yield to Conversion (%)
Total Conversion (%)
100
60
0
(a)
0.25
0.5
0.75
Residence Time (min)
2.0 Amine equiv.
90
4.0 Amine equiv.
1.2 Amine equiv.
80
70
60
0
1
(b)
0.25
0.5
0.75
1
Residence Time (min)
Figure 5.8. Production of metoprolol 20 at various amine 19 ratios: (a) conversion of epoxide 18; (b)
fraction of conversion comprised of desired product 20.
119
Application to indacaterol.
The adaptation of the indacaterol aminolysis to a microreactor system presented
several challenges. First, the reported reaction time in diglyme at elevated temperatures
was approximately 15 h (Scheme 5.3).150 Such lengthy residence times are not possible in
a microreactor system due to difficulties in delivering the fluid in a stable (non-pulsating)
manner using syringe pumps at such low flow rates. As discussed in section 5.2, attempts
to catalyze this reaction with a variety of known aminolysis promoters ultimately did not
lead to reaction times that were amenable to microreactors. However, simply heating at
elevated temperatures in polar protic solvents such as ethanol resulted in reaction times
that could be considered in microreactors (approx. 30 min).
The second obstacle to performing the indacaterol aminolysis in the microreactor was
low solubility of the starting epoxide 13 in commonly used solvents. The quinolinone
structure provided a highly crystalline material that had a limited solubility (<0.1 M) in
most organic solvents, including ethanol and acetonitrile. Formation of solids in the
microreactor often clogged the inlets, preventing flow. To solve this problem, a solvent
screen was conducted. N-Methylpyrrolidone (NMP) was found to provide reasonable
solubility of 13 (~0.5 M), and the dielectric constant of NMP is similar to that of
acetonitrile. We were also able to keep the concentration of the reaction high by premixing the amine and epoxide to flow the mixture from one syringe. This technique
avoids further dilution of the reaction when the two components are introduced
separately, thus enabling higher overall conversion.
The reaction rate at room
temperature is insignificant, eliminating concerns of product formation in the syringe.
The formation of 15 was also limited by its thermal stability as a free base; it has been
reported that 15 is unstable in organic solvents.150 Indeed, we observed significant
decomposition when the indacaterol precursor was heated to temperatures above 200ºC.
Considering these issues, a solution of 13 and 14 was prepared in NMP, and 10% water
was added as a promoter for the aminolysis reaction, based on previous reports of water
as a promoter of epoxide aminolysis.132
Approximately 3% conversion to 15 was
obtained in the syringe after 12 h at room temperature; this was considered an acceptable
margin. Initially, a 0.4 M solution was pumped through the microreactor at 185ºC and
varying flow rates to determine the reaction parameters. Excellent conversion (97%) was
120
obtained at 185ºC in only 15 min with 68% of the desired indacaterol precursor 15
produced.
Yields and selectivities observed under microreactor conditions mirrored
those obtained by heating in diglyme for 15 hours.150 Small amounts of 13 were found to
have crystallized out in the syringe after 12 h but did not lead to crystallization or
clogging in the microreactor.
At a slightly decreased concentration of the starting
solution (0.38 M of 13), 13 was completely soluble, and performing the aminolysis
reaction in quadruplicate under the same conditions led to only minor variation in the
yield of 15 (minimum of 68, maximum of 70%). Figure 5.9 shows the results of this
reaction at 185ºC with amounts of water and concentrations.
100
Conversion (%)
80
60
Product
Bisalkylation
Total
40
20
0
0
5
10
15
Residence Time (min)
20
Figure 5.9. Aminolysis to form indacaterol precursor 15 in a microreactor at 185ºC and 250 psi;
open and closed symbols represent NMP/8% H2O with 0.38 M of 13 and NMP/10% H2O with 0.4 M
of 13, respectively. Regioisomer formation (not shown) follows a trend similar to that of
bisalkylation.
Decreasing the temperature to 165ºC reduced the degree of thermal decomposition of
15, and slightly increased yields were obtained at the expense of longer reaction times.
Similarly, increasing the temperature to 200ºC led to better conversion at shorter times at
the expense of overall product yield due to thermal decomposition. Under the best
observed conditions (NMP/10% H2O, 185ºC, 0.38 M of 13), 1.5 g/d (0.5 kg/year) of the
indacaterol precursor 15 could be obtained from a single 120-µL microreactor.
121
5.5
Kinetic Evaluation of Epoxide Aminolysis
To enable the determination of the optimal set of concentrations and conditions for
performing epoxide aminolysis, a systematic evaluation of the kinetic parameters of the
reaction is necessary. As discussed in section 5.4.3, the aminolysis of styrene oxide 5
with 2-aminoindan 6 was applied as a model chemistry that is electronically similar to the
formation of indacaterol precursor 15. The kinetic evaluation was performed using
ethanol as the solvent. To fully determine the rate of formation of the desired product 7
(Scheme 5.2), the kinetics of the bisalkylation reaction (aminolysis of SO 5 with product
7 to form side products 9 and 10) were first investigated. The determined reaction rate
law of bisalkylation was then applied to decouple the kinetics of the primary reaction and
to obtain the overall rate law of product 7 formation.
5.5.1 General Procedure
The setup to perform the kinetics determination was that described in section 5.3.2
using three syringe pumps and a 250-psi backpressure regulator. The applied procedure
was nearly identical to that for reaction screening and profiling using the microreactor
system, as described in section 5.4.1, but with all three flow rates (epoxide solution,
amine solution, and pure solvent) being varied to obtain different concentrations and
stoichiometries from a single prepared reagent solution set. Similarly to the reaction
profiling procedure, five residence times were allowed to pass following establishment of
new conditions. At each set of conditions, samples were collected in triplicate. HPLC
analysis was used for reactant and product quantification, with ~10 mol% of naphthalene
(vis. SO 5) as the internal standard, using the same method as previously. Nearly all
samples had mass balances between 96% and 102%, which was judged acceptable based
on HPLC variability. The few samples with mass balances outside this range were not
considered in the kinetics evaluation.
To perform the evaluation of the bisalkylation kinetics, it was necessary to synthesize
7, which was performed using the microreactor system. Two syringes were loaded with 2
M SO 5 and 2.4 M AI 6, respectively, each in ethanol (resulting in 1 M and 1.2 M of 5
and 6, respectively, in the reactor). The solutions were then flowed into the microreactor
122
at 225ºC and 500 psi with a residence time of 4 min, and the outflow was collected.
Upon cooling to room temperature and solvent evaporation, an off-white crystalline solid
was formed.
1
H-NMR and HPLC confirmed it to be the desired product 7 with 97%
purity (the balance was side product 8). This was deemed acceptable for the kinetic study
and was used without further purification.
The utility of microreactors for kinetic studies was highly evident here. It was
possible, with a single preparation of two reagent solutions, to scan up to 35 sets of
conditions (concentrations, temperatures, and residence times) with samples in triplicate,
generating over 100 data points within one 8-hour period. Additionally, this procedure
minimizes reactant use and waste, as only 0.5 to 2 g of each reagent was necessary for
such an experiment set, and of the total of 30 mL of reaction solution (including solvent
for concentration control), fully 10 mL was used directly as HPLC samples.
5.5.2 Kinetic Evaluation of Bisalkylation
Upon reacting SO 5 with 7, two product peaks were observed by HPLC,
corresponding to two diastereomers of 9 (resulting from using a racemic mixture of SO
5). This was confirmed by performing an experiment with an optically pure enantiomer
of SO 4 and observing only one bisalkylation peak. No peak corresponding to 10 was
observed, indicating nearly 100% selectivity of the bisalkylation towards regioisomer 9.
The order of the reaction of the formation of 9 from SO 5 and 7 was determined by
analyzing the results of varying reagent concentrations (Figure 5.10), as well as fitting all
obtained data points to a rate model of first order in each of SO 5 and 6 (designated as
Pr), plotting
⎡⎛ [Pr]0 − [SO]0 ⎞ ⎛ [Pr]0 ⎞ ⎤
1
1
1
ln ⎢⎜1 −
−
⎟ ⎥ versus time or
⎟⎜
[SO]0 − [Pr]0 ⎣⎢⎝
[Pr]
[Pr] [Pr]0
⎠ ⎝ [SO]0 ⎠ ⎦⎥
versus time for equivalent starting amounts of SO 5 and 7. The mass balances of the
sample analysis closed to within the aforementioned range relative to both of the reagents
(SO 5 and 7). The amount of initial regioisomer 8 remained unchanged (to the limit of
our detection) in all samples, indicating that the rate of aminolysis of SO 5 by this amine
is insignificant and can be disregarded in the overall rate law estimation. Additionally,
no peaks corresponding to 10 or 11 were observed.
123
-3
2.0×10
(a)
1.6×10
-3
1.2×10
-3
[SO]_0=0.025 M; [Pr]_0=0.025 M
[SO]_0=0.025 M; [Pr]_0=0.050 M
[SO]_0=0.025 M; [Pr]_0=0.10 M
[SO]_0=0.050 M; [Pr]_0=0.050 M
Concentration (mol/L)
Concentration (mol/L)
2.0×10
8.0×10 -4
4.0×10
-4
-3
(b)
1.6×10
-3
1.2×10
-3
[SO]_0=0.025 M; [Pr]_0=0.025 M
[SO]_0=0.025 M; [Pr]_0=0.050 M
[SO]_0=0.025 M; [Pr]_0=0.10 M
[SO]_0=0.050 M; [Pr]_0=0.050 M
8.0×10 -4
4.0×10
-4
0
0
0
50
100
150
200
250
0
50
100
Time (s)
150
200
250
Time (s)
ln[f([Pr])]
3
R2 = 0.9911
2
‐3 ‐1 ‐1
k = (6.14±0.5)x10 M s
1
0
(c)
0
100
200
300
Time (s)
Figure 5.10. Bisalkylation reaction order determination through evolution of concentrations of the
two diastereomers of product 9 [(a)and (b)] (◊ : initial concentrations of SO 4 and of 7 of 0.025 M
each; □ : initial concentration of SO 4 of 0.025 M and of 7 of 0.050 M; Δ : initial concentration of SO
4 of 0.050 M and of 7 of 0.10 M; ○ : initial concentrations of SO 4 and of 7 of 0.050 M each); (c) plot
of all reaction data points as a natural logarithm function of concentration of 7 vs. time.
The initial rates of reaction were approximated by measuring the amount of product
produced at the 20-second residence time. For the baseline experiments with initial
concentrations of 0.025 M each of SO 5 and of 7, the initial rates were measured to be
6.37×10-6 mol L-1 s-1 and 4.56×10-6 mol L-1 s-1 for the two diastereomers of 9. When the
initial concentration of 7 was doubled to 0.050 M, keeping the concentration of SO 5 the
same, the initial rates were measured to be 1.43×10-5 mol L-1 s-1 and 1.09×10-6 mol L-1 s-1
for the same two diastereomers of 9, increasing by factors of 2.2 and 2.4, respectively.
When the initial concentration of SO 5 was also doubled (both reagents now initially at
0.050 M), the initial rates increased to 2.96×10-5 mol L-1 s-1 and 2.36×10-6 mol L-1 s-1 f,
further increasing by factors of 2.1 and 2.2, respectively. Similar increases over the
second case were observed with concentrations of SO 5 and 7 of 0.025 M and 0.10 M,
respectively. Additionally, the plot of Figure 5.10c fit well to a linear correlation.
124
Combined, these results indicate that, at the evaluated conditions, the reaction is firstorder with respect to each of SO 5 and 7, or following the rate law:
rBis = rBis1 + r Bis 2 = k Bis1[SO][Pr] + k Bis 2 [SO][Pr] = ( k Bis1 + k Bis 2 ) [SO][Pr]
5.8
where Pr is desired product 7 and kBis1 and kBis2 represent the rate constants of
bisalkylation to form the two diastereomers of 9.
-4.6
ln kbis (M-1 s-1)
-4.8
Ea/R = -1014±281 K
-5
-5.2
Bisalkylation 1
Bisalkylation 2
Ea/R = -1445±271 K
-5.4
-5.6
-3
2.2×10
-3
-3
2.3×10
2.4×10
-3
2.5×10
Inverse Temperature (K-1)
Figure 5.11. Arrhenius correlation between temperature and rate constants of bisalkylation.
The Arrhenius dependence was performed by varying the temperature between 140ºC
and 180ºC, at a residence time of 60 s and concentrations of SO 5 and 7 of 0.05 M each.
Figure 5.11 shows the temperature dependences of the formation of two diastereomers of
9 as plots of ln(kBis1) and ln(kBis2) vs. 1/T. The Arrhenius parameters were determined as:
k Bis1 = e
⎛ 8.4 ± 2.4 kJ / mol ⎞
( −2.66 ± 0.65) − ⎜
⎟
RT
⎝
⎠
k Bis 2 = e
k Bis1,453 K = (7.5 ± 6.9) ×10−3 M −1s −1
⎛ 12.0 ± 2.2 kJ / mol ⎞
( −1.89 ± 0.62) − ⎜
⎟
RT
⎝
⎠
5.9
k Bis 2,453 K = (6.2 ± 5.3) × 10−3 M −1s −1 5.10
The confidence intervals in the above equations were determined using the error
propagation method.168 For a function f(a,b,c…), with known standard deviations of the
parameters a, b, c, etc., the standard deviation of f can be calculated as follows:
2
2
⎛ ∂f ⎞
⎛ ∂f ⎞
σ = ⎜ ⎟ σ a2 + ⎜ ⎟ σ b2 + …
⎝ ∂a ⎠
⎝ ∂b ⎠
2
f
5.11
assuming that each parameter error is small and evenly distributed about the mean.
125
A rate constant k with Arrhenius functionality thus has the following error function:
2
2
⎛ ∂k ⎞ 2 ⎛ ∂k ⎞ 2
⎛ ∂k ⎞ 2
Ea2
1
2⎛ 2
2
2⎞
σ =⎜
⎟ σ Ea + ⎜
⎟ σ ln A + ⎜
⎟ σ T = k ⎜ σ A + 2 2 σ Ea + 2 4 σ T ⎟ 5.12
RT
RT
⎝ ∂T ⎠
⎝ ∂ (ln A) ⎠
⎝
⎠
⎝ ∂Ea ⎠
2
2
k
A conservative temperature variation of 1ºC was assumed (see section 5.3.3).
5.5.3 Kinetic Evaluation of Primary Aminolysis
Having the information regarding the kinetics of bisalkylation enabled us to account
for the rate of consumption of SO 5 due to that reaction in addition to the primary
aminolysis. At the explored conditions, the ratios of the two bisalkylation products were
nearly identical to the ratios obtained at the same temperature during the bisalkylation
kinetics investigation (section 5.5.2), and no quantifiable amounts of bisalkylation
products 10 or 11 were observed by HPLC. This indicates that the side product 8 does
not participate in the bisalkylation reaction to a significant extent, thus allowing us to
discount the consumption of SO 5 by that reaction route.
The orders of the reaction of the formation of 7 and 8 from SO 5 and AI 6 were
determined by analyzing the results of varying reagent concentrations (Figure 5.12), as
well as fitting all obtained data points to a rate model of first order in each of SO 5 and
AI 6, plotting
⎡⎛ [AI]0 − [SO]0 ⎞ ⎛ [SO]0 ⎞ ⎤
1
ln ⎢⎜1 +
⎟ ⎥ versus time. Based on the
⎟⎜
[AI]0 − [SO]0 ⎢⎣⎝
[SO]
⎠ ⎝ [AI]0 ⎠ ⎦⎥
above discussion, the sum total of the bisalkylation products was attributed to the reaction
of 7 with SO 5; thus, the produced concentrations of 9 and 10 were added to that of 7 for
the calculation of the rate constants.
The initial rates of reaction were approximated by measuring the amount of product
produced at the 20-second residence time. For the baseline experiments with respective
initial concentrations of 0.10 M and 0.12 M of SO 5 and AI 6, the initial rates were
measured to be 2.86×10-4 mol L-1 s-1 and 8.67×10-4 mol L-1 s-1 for 7 and 8, respectively.
When the initial concentration of AI 6 was doubled to 0.24 M, keeping the concentration
of SO 5 the same, the initial rates were measured to be 5.85×10-4 mol L-1 s-1 and 1.83×104
mol L-1 s-1 for 7 and 8, increasing by factors of 2.0 and 2.1, respectively. When the
initial concentration of SO 5 was also doubled, to 0.20 M, the initial rates increased to
126
1.10×10-3 mol L-1 s-1 and 3.49×10-3 mol L-1 s-1 for 7 and 8, both further increasing by a
factor of 1.9. Similar increases over the second case were observed with concentrations
of SO 5 and AI 6 of 0.10 M and 0.48 M, respectively.
3.0×10-2
(a)
[SO]_0=0.10 M; [AI]_0=0.12 M
[SO]_0=0.10 M; [AI]_0=0.24 M
[SO]_0=0.10 M; [AI]_0=0.48 M
[SO]_0=0.20 M; [AI]_0=0.24 M
[SO]_0=0.20 M; [AI]_0=0.48 M
0.08
0.06
0.04
0.02
2.5×10-2
Concentration (mol/L)
Concentration (mol/L)
0.10
[SO]_0=0.10 M; [AI]_0=0.12 M
[SO]_0=0.10 M; [AI]_0=0.24 M
[SO]_0=0.10 M; [AI]_0=0.48 M
[SO]_0=0.20 M; [AI]_0=0.24 M
[SO]_0=0.20 M; [AI]_0=0.48 M
(b)
-2
2.0×10
1.5×10-2
1.0×10-2
0.5×10 -2
0.00
0
0
50
100
150
0
Time (s)
50
100
150
Time (s)
7
ln[f([SO])]
6
R2 = 0.9987
5
4
k = (2.12±0.05)x10‐2 M‐1s‐1
3
2
1
0
0
(c)
100
200
300
Time (s)
Figure 5.12. Aminolysis reaction order determination of (a) product 7 and (b) regioisomer 8 by
concentration variation (◊ : initial concentrations of SO 5 and AI 6 of 0.10 M and 0.12 M,
respectively; □ : initial concentrations of SO 5 and AI 6 of 0.10 M and 0.24 M, respectively; Δ : initial
concentrations of SO 5 and AI 6 of 0.10 M and 0.48 M, respectively; ○ : initial concentrations of SO 5
and AI 6 of 0.20 M and 0.24 M, respectively; × : initial concentrations of SO 5 and AI 6 of 0.20 M and
0.48 M, respectively); (c) plot of all reaction data points as a natural logarithm function of
concentration of SO 5 vs. time.
The plot of Figure 5.12c fit very well to a linear correlation. Combined with the
initial rate analysis, these results indicate that, at the evaluated conditions, both
bisalkylation reactions are first-order with respect to each of SO 5 and AI 6, or following
the rate law:
ri = ki [SO][AI]
5.13
where ri represents the rate of formation of 7 (k1) or of 8 (k2). Therefore, the rates of
formation of the product 7 and of consumptions of SO 5 and AI 6 are expressed as
follows:
127
rPr = k1[SO][AI] − ( k Bis1 + k Bis 2 ) [SO][Pr]
5.14
rSO = − ( k1 + k2 ) [SO][AI] − ( k Bis1 + k Bis 2 ) [SO][Pr]
5.15
rAI = − ( k1 + k2 ) [SO][AI]
5.16
where kBis1 and kBis2 were defined and calculated in the previous section.
-3.6
-4
ln kbis (M-1 s-1)
Ea/R = -3277±241 K
-4.4
-4.8
Ea/R = -1946±219 K
-5.2
-5.6
-3
2.2×10
-3
-3
2.3×10
2.4×10
Product
Regioisomer
-3
2.5×10
Inverse Temperature (K-1)
Figure 5.13. Arrhenius correlation between temperature and the rate constants of SO 5 aminolysis.
The Arrhenius dependence was performed by varying the temperature between 140ºC
and 180ºC, at a residence time of 60 s and concentrations of SO 5 and AI 6 of 0.10 M and
0.12 M, respectively.
Figure 5.13 shows the temperature dependences of 7 and 8
formation as plots of ln(k1) and ln(k2) vs. 1/T.
The Arrhenius parameters were
determined to be as follows, with errors calculated as described in the previous section:
k1 = e
⎛ 27.2 ± 2.0 kJ / mol ⎞
(3.41± 0.55) − ⎜
⎟
RT
⎝
⎠
k2 = e
⎛ 16.2 ±1.8 kJ / mol ⎞
( −0.80 ± 0.50) − ⎜
⎟
RT
⎝
⎠
k1,453 K = (2.2 ± 1.7) ×10−2 M −1s −1
5.17
k2,453 K = (6.1 ± 4.2) ×10−3 M −1s −1
5.18
As expected based on the electron properties of the amine nitrogen, which make it
more reactive as a secondary amine than as a primary amine,169 the activation energies of
the primary alkylation are higher than those of the bisalkylation. However, the preexponential factors are much greater, as would be expected based on the steric effects.
128
Overall, this indicates that, while the bisalkylation is often much faster than the first
alkylation in similar reactions at lower temperatures, the large disparity in activation
energies leads to a much better selectivity for the desired single alkylation at increased
temperatures. Additionally, the slightly higher activation energy towards the desired
product 7 as compared to the regioisomer 8 also leads to improved selectivity for the
desired product with increased temperature. The fairly similar activation energies of the
primary aminolysis and bisalkylation indicate that the product distribution would only be
a weak function of temperature, which has been observed in our profiling experiments.
The conditions used during the reaction screening experiments with SO 5 and AI 6
have been inserted into the model given by equations 5.14 through 5.16. Figure 5.14
shows the comparison between the model calculated reaction yield with time and the
experimentally obtained values. The slight deviations between the calculated model
trend and the actual data may be due to experimental error such as the use of older AI 6,
which tends to oxidize, degrading with age, leading to lower concentration in the stream.
The model can also be used to provide a response surface of product yield with varying
parameters such as reaction temperature, residence time, and concentrations and
stoichiometric ratios of reagents. Figure 5.15 shows some of the time slice cross-sections
of the response surface, demonstrating the effects of the aforementioned parameters.
Product Yield (%)
80
60
40
Experimental
20
Model
0
0
1
2
3
4
Residence Time (min)
5
Figure 5.14. Comparison of experimental and calculated yields for the aminolysis of SO 5 with AI 6.
129
0.6
(a)
(b)
0.8
0.5
0.7
0.4
0.6
`
140ºC
160ºC
180ºC
200ºC
220ºC
0.2
0.1
Yield
Yield
0.5
0.3
0.4
`
[SO]_0=0.1 M; [AI]_0=0.24 M
0.3
[SO]_0=0.2 M; [AI]_0=0.24 M
0.2
[SO]_0=0.4 M; [AI]_0=0.48 M
0.1
[SO]_0=0.4 M; [AI]_0=1.2 M
0
[SO]_0=1.0 M; [AI]_0=1.2 M
0
0
100
200
300
Time (s)
400
500
600
0
100
200
300
Time (s)
400
500
600
Figure 5.15. Modeled yield of 7 with time: (a) at different temperatures with starting concentrations
of 0.2 M SO 5 and 0.24 M AI 6; (b) with different initial reagent concentrations at 180ºC.
Because the rate constant of formation of 7 has the strongest dependence on
temperature among the calculated rate constants, the model shows that higher
temperatures lead to an improved overall yield in addition to a faster reaction. Indeed,
we have seen that to be the case in the reaction screening study (Table 5.1). Additionally,
the model shows that a higher excess of the amine leads to a better yield relative to the
epoxide. Because the amine is often a much less expensive reagent and can also be
recovered after the reaction, it may be desirable to perform this reaction on scale at very
high excesses of amine to drive the highest possible product yield. If a smaller excess of
amine is used, then there may be a maximum in the yield relative to time, as there may be
a point at which there is sufficient product and epoxide in solution for the bisalkylation
reaction to be faster than the primary product formation. Therefore, if it is desirable to
use only a small excess of the amine, then it is important to know when said yield
maximum occurs and to stop the reaction at that point, rather than proceeding to full
conversion of the epoxide, as this would only cause loss of product. This underscores the
significance of understanding the reaction kinetics.
5.6
Reaction Scale-up
To demonstrate the applicability of the microreactor-obtained kinetic knowledge, the
derived kinetic model for the aminolysis of SO 5 with AI 6 was applied to scaled-up flow
and concentrations of reagents. The increased production was intended to verify that the
information resulting from microscale experiments is applicable to meso-scale systems,
from which, performance in macroscale plants can be gauged more easily.
130
Based on the kinetic model derived in section 5.5, a set of conditions was selected.
As can be seen in Figure 5.15, the optimal yield relative to the epoxide reagent is
achieved at higher temperatures and with larger excess of the amine reagent. Thus, the
selected conditions for this reaction were as follows. The concentration of SO 5 was
selected as 0.4 M because this represents the solubility limit for the indacaterol precursor
epoxide in section 5.4.4, the reaction which this model is intended to mimic. The
concentration of AI 6 was selected as 1.2 M, or a factor of 3 of the epoxide concentration.
As any excess amine is to be recovered later in the indacaterol process, this is feasible
both economically and in the process setup.
The reactor selected for the epoxide aminolysis was a stainless-steel tube. Because
this reaction is performed at high pressures, it was not feasible to perform it in silicon
meso-reactors, as such reactors are not as robust to pressure as their microscale
counterparts due to a much smaller relative bonded area (see Chapter 2). However,
despite the lack of reaction visibility, the smoothness of the 316 stainless steel tube is
expected to allow some degree of superheating, as there are no ready nucleation sites and
no solids formed within the reaction medium. The reaction temperature was thus selected
to be 220ºC, which is slightly above the boiling point of ethanol at 34 bar (206ºC), the
highest backpressure applied in the screening and kinetics studies.
The reactor was a 6-ft-long (1.8 m), 316 stainless-steel smooth-bore seamless tube,
¼” OD, 0.12” ID (3 mm) (McMaster, 89785K52), coiled at a 10-cm turn diameter, with
171 cm (12.5 mL) submerged in a 2-liter high-temperature silicone oil 200.50 (Hart
Scientific, 5014). The reactor was connected on both ends to 1/16” stainless-steel tubing
by stainless-steel reducing swaging unions (McMaster, 5182K242). At the outlet end, 30
cm of narrow tubing connected the reactor to the 500-psi backpressure regulator used in
previous studies, which flowed out to a collection bottle. At the inlet end, 20 cm of
narrow tubing connected to the reactor a compression-packaged second-generation twostream micromixer described in Chapter 3. The two inlets of the micromixer were fed by
HPLC single-piston pumps (Rainin Dynamax SO-200). The oil bath was placed on a
stirplate with a 10-cm-long magnetic stirbar, stirred at 200 rpm.
The heating was
achieved by an immersion heater controlled by a J-KEM Scientific 2-channel controller,
monitored by a K-type thermocouple probe. The outer walls of the oil bath were covered
131
with 1”-thick fiberglass rope for thermal insulation.
At 220ºC, placing another
thermocouple at different positions within the bath (including by the bath wall vs. center,
at the bottom vs. near the surface) showed a maximum temperature difference of 0.2ºC.
Using the conditions described above and the derived kinetic model, 99% conversion
of SO 5 is expected at 110 seconds, resulting in 76.0% yield. To calculate the confidence
intervals of this value, a finite difference approach was taken. From equations 5.14-5.16,
the concentrations of the starting materials and the desired product (Pr) can be evaluated
as follows:
[SO]i = [SO] i −1 − ( ( k1 + k2 ) [SO]i −1[AI]i −1 + ( k Bis1 + k Bis 2 ) [SO]i −1[Pr]i −1 ) ( ti − ti −1 ) 5.19
[AI]i = [AI] i −1 − ( ( k1 + k2 ) [SO]i −1[AI]i −1 ) ( ti − ti −1 )
5.20
[Pr]i = [Pr] i −1 + ( k1[SO]i −1[AI]i −1 − ( k Bis1 + k Bis 2 ) [SO]i −1[Pr]i −1 ) ( ti − ti −1 )
5.21
Therefore, error will propagate throughout the time interval of integration as follows:
2
2
2
⎡
⎤
σ SO,
i = σ SO,i −1 ⎣1 − ( ti − ti −1 ) ⎡
⎣( k1 + k2 ) [AI]i −1 + ( k Bis1 + k Bis 2 ) [Pr]i −1 ⎤⎦ ⎦ +
{
2
2
2
2
... ( ti − ti −1 ) σ AI,
i −1 ⎡
⎣( k1 + k2 ) [SO]i −1 ⎤⎦ + σ Pr,i −1 ⎡⎣( k Bis1 + k Bis 2 ) [SO]i −1 ⎤⎦ + ...
(
... + σ k21 + σ k22
) ([SO]
i −1[AI]i −1
)2 + (σ k2
Bis1
+ σ k2Bis 2
) ([SO]
i −1[Pr]i −1
5.22
)2 }
2
2
2
σ AI,
i = σ AI,i −1 ⎡
⎣1 − ( ti − ti −1 ) ( k1 + k2 ) [SO]i −1 ⎤⎦ + ...
{
2
(
2
2
2
... ( ti − ti −1 ) σ SO,
i −1 ⎡
⎣( k1 + k2 ) [AI]i −1 ⎤⎦ + σ k1 + σ k2
) ([SO]
i −1[AI]i −1
)2
}
5.23
2
2
2
σ Pr,
i = σ Pr,i −1 ⎡
⎣1 + ( ti − ti −1 )( k Bis1 + k Bis 2 ) [SO]i −1 ⎤⎦ +
{
2
2
2
⎡
⎤
... ( ti − ti −1 ) σ SO,
⎡k1[SO]i −1 ⎦⎤ + ... 5.24
i −1 ⎣( k1[AI]i −1 − ( k Bis1 + k Bis 2 ) [Pr]i −1 ) ⎦ + σ AI,i −1 ⎣
2
(
... + σ k21 ([SO]i −1[AI]i −1 ) + σ k2Bis1 + σ k2Bis 2
2
) ([SO]
i −1[Pr]i −1
)2 }
Using the k values and their standard deviations given in equations 5.9-5.10 and 5.175.18, as well as assuming a 1% error in initial concentrations of starting materials (a
value to which the overall error had almost no sensitivity), the error propagation for the
three species’ concentrations was obtained. As the yield is simply the ratio of the final
concentration of Pr 7 to the starting concentration of SO 5, the standard deviation of yield
is equal to the ratio of the standard deviation of the final concentration of Pr 7 to the
starting concentration of SO 5 (the 1% error in [SO]0 is negligible in the calculations). At
132
the conditions selected for scale-up, the confidence interval of yield at 120 s residence
time was calculated to be 4.9%.
However, these values are obtained assuming plug flow, which was reasonable in the
microreactor (the axial dispersion Péclet number PeAx given by equation 2.4 ranged from
600 down to 100). As the larger-scale reactor has a fairly large inner diameter and as
flow will be fairly rapid (6.24 mL/min for the 120 s residence time selected), dispersion
is expected to play a large role. At the flow rate of 6.24 mL/min, using the diffusion
coefficient D of 10-9 m2/s (while exact values for the starting materials in ethanol were
not available, compounds similar in size and functional groups displayed diffusivities of
this scale170), the dispersion coefficient D (given by equation 2.3) was found to be
9.8×10-3 m2/s, with PeAx of 2.6, representing significant dispersion. Using this dispersion
coefficient, the first moment of residence time distribution E was calculated according to
equation 2.5 and is shown in Figure 5.16. The conversion in a reactor with dispersion is
given as:52
∞
X out = ∫ X A,PFR (t )E(t )dt
5.25
0
5×10 -3
4×10 -3
E(t)
3×10 -3
2×10 -3
1×10 -3
0
0
500
1000
1500
2000
Time (s)
Figure 5.16. First moment of residence time distribution calculated using dispersion in a straight
steel tube reactor.
133
Performing this calculation on the kinetic model results in a yield of the product 3 of
73.0%±4.7%, about 3.3% lower than what is expected in a plug flow reactor, although
the confidence intervals overlap with those of yield without dispersion. Additionally,
because it is a curved tube, dispersion is expected to be reduced due to Dean flow. In
Dean flow, fluid is transported from the inner to the outer wall due to the balance of
forces on the fluid caused by the curvature of the tube, as shown in Figure 5.17, adapted
from Sudarsan and Ugaz.173 This transport of fluid across the cross-section greatly
increases radial mixing, which acts to counter the effects of dispersion due to the
parabolic flow profile. The Dean number De, given as:
De = Re⋅
D
2R
5.26
where D is the inner diameter of the pipe and R is the curvature radius, was calculated to
be 4.2. With the Schmidt number (using properties of pure ethanol) calculated to be
1500, this gives a value of De2Sc of 26500, in which regime, the effective dispersion is
expected to be about 20% of the calculated value,174 making it more similar to plug flow.
Thus, the expected yield of 7 would be 75.4%±4.9%. Overall, the yields with and
without dispersion and Dean flow have overlapping confidence intervals; thus, it is not
expected to be able to determine experimentally the effect of dispersion on this system.
Figure 5.17. Cartoon of the effect of Dean flow in a curved cylindrical tube on two incoming sections
of fluid; the bottom array demonstrates the effect of increasing Dean number (from left to right).
Adapted from Sudarsan and Ugaz.173
134
This reaction was performed by flowing a total of 163 mL (13 residence volumes) of
solution through the reactor, taking a total of less than 30 minutes. Samples were taken at
3, 8, and 12 residence volumes. Each sample showed 100% conversion (no significant
amount of SO 5 was detected), and the yield of the desired product 7 was between 78.0
and 78.2% for all three samples (performed using the same HPLC analysis as discussed
in section 5.5.1. The yield is slightly higher than predicted by the kinetic model using the
average parameters; however, it is well within what is predicted by the model if the
parameter confidence intervals are considered.
Overall, the meso-scale setup
reproducibly synthesized 9.2 g of desired product 7 in 30 minutes, with a higher yield
than previously reported for this reaction.
5.7
Conclusion
The aminolysis of epoxides using a continuous-flow microsystem was shown to be a
highly efficient process compared with traditional batch synthesis. The high pressures
permitted by the microreactor allowed for heating of solutions to temperatures of up to
245ºC, leading to excellent yields and conversions of simple terminal epoxides at
residence times under 5 min in ethanol. The aminolysis of more sterically hindered
epoxides also proved successful, although longer reaction times were necessary.
Additionally, due to the elimination of headspace as compared to batch, volatile amines
can be used in the reaction at precise concentrations, without concern of partitioning into
the vapor phase. The use of a small amount of a polar protic solvent to accelerate the
aminolysis reaction can also be applied without concern for the volatility of the solvent
components.
Application of epoxide aminolysis in a continuous-flow microreactor
towards the production of metoprolol led to product outputs of 7.0 g/h in a single
microreactor system. In a more challenging example, the final precursor of indacaterol
was also produced by this method at a residence time that was a factor of 60 shorter than
that reported in the literature, with similar yield and product distribution.
The kinetics of a model chemistry simulating indacaterol precursor synthesis was
investigated, using styrene oxide 5 and 2-aminoindan 6 as the reagents. The bisalkylation
reactions between the desired product 7 and the epoxide were investigated individually
and decoupled from the primary aminolysis kinetics. The overall reaction model for the
135
production of the desired product was obtained, including the temperature dependence.
The model was found to fit well with previously obtained experimental data. Overall, the
model suggests performing this reaction at the highest possible temperatures with a large
excess of the amine reagent to obtain optimal yields. If only a small excess of amine is
used, there may be a maximum of product yield with time, following which, yield begins
to decrease due to the bisalkylation reaction.
This study demonstrated the utility of microreactor-based flow systems for
reaction acceleration, profiling, and kinetic study.
A robust high-pressure, high-
temperature microreactor with very rapid thermal control enabled highly efficient
reaction and kinetics evaluations, greatly reducing reagent consumption and experiment
time. A total of less than 5 g each of styrene oxide 5 and 2-aminoindan 6 were necessary
to perform a full multi-step kinetic study, with the ability to complete up to 35 reactions
in a wide range of concentrations, residence times, and temperatures in a single workday.
As more complex pharmaceutical precursors can be highly expensive and difficult to
synthesize, the ability to perform a screen and a kinetics study with only grams of reagent
and in a short time period can be highly valuable to the fine chemicals industries. The
determined kinetic parameters indicated that the activation energy of the formation of the
desired product 7 is higher than those for the regioisomers formation and for the
bisalkylation reactions. This results in the fortuitous effect that conditions that lead to a
faster reaction and higher conversion (higher temperature) also improve reaction
selectivity towards the desired product. A scale-up of this reaction was performed in a
12.5-mL steel tube meso-reactor, for which dispersion was calculated and shown to have
very little effect. Synthesis of 18 g/hr of desired product 7 was achieved, with 100%
conversion and 78% yield, in good agreement with the kinetic derivations.
136
Chapter 6.
Solids Handling in Flow†
Silicon microfluidic systems have been demonstrated to be useful tools for reaction
screening, profiling, and optimization due to the rapid thermal equilibration, capability of
wide ranges of conditions, and chemical compatibility. For this reason, microreactor
systems are appealing as study tools for industrially interesting reactions. However, a
large majority of such reactions involve solids, either as a reactant, the desired product, or
a side product. One of the predominant challenges of flow chemistry is the formation and
flow of solids, which results in constrictions within the flow path, ultimately leading to
blockage.
Therefore, to enable the application of microreactors as study tools for
reactions involving solids, it is necessary to understand how solids form into blockages in
microchannels and to apply that understanding to a system designed to allow flow
containing solids.
To study solids formation in flow, we selected as a model solids-generating reaction
the palladium-catalyzed C-N coupling of an aryl chloride with aniline, forming insoluble
NaCl salt in dioxane. It was observed that solids formed into blockages due to two
primary mechanisms: surface deposition (constriction) and bulk agglomeration
(bridging).
By applying reactor design, surface modification, and high-frequency
acoustics (ultrasonication), the two clog formation mechanisms were studied in more
detail for a fuller understanding of the blockage process. A set of reactors was designed
to combine techniques such as gradual turn layout, concentration control, surface
modification, and ultrasonication while taking advantage of silicon microreactor
properties (compactness, robustness, ease of visualization of flow). With the gained
understanding of solid clog formation, progress has been made towards enabling flow of
solids-generating reactions in microchannels.
†
This chapter describes work done in close collaboration with Ryan L. Hartman, who at
the time was a post-doctoral researcher in the laboratory of Prof. Klavs F. Jensen, and
John R Naber, who at the time was a doctoral student in the laboratory of Prof. Stephen
L. Buchwald in the Department of Chemistry at MIT.
137
6.1
Introduction
The majority of chemical reactions involved in the syntheses of fine chemicals such
as pharmaceuticals, cosmetics, food additives, and agricultural products require
multiphase interactions, including gas-liquid, liquid-liquid, and solid-liquid.146,
173, 174
Microreactor utility has been demonstrated for reaction study by enhancing mass and heat
transfer,7, 84, 143, 157, 175-183 including the study of mass transfer coefficients in multiphase
systems,84, 157, 175, improved reaction control,176-179, and greatly reduced waste.143, 180
Interest in the ability to handle solids in microfluidic systems has been steadily
growing in recent years because of increasing demand for better studies of heterogeneous
operations, including the manipulation of biological materials and chemical
transformations. To enable the design and assembly of a robust system capable of a wide
array of solids-generating or reacting-solid syntheses, a fundamental understanding of
how solids clog microsystems is necessary. Such understanding will greatly aid in the
development of strategies to overcome the challenge of slurry flowability on the
microscale, allowing us to apply microsystems to a broader range of reaction discovery
and development. Microchemical syntheses can benefit from enhanced heat and mass
transfer characteristics, safety of operation, isolation of sensitive reactions from air or
moisture, improved reaction screening, reduction of hazardous waste, and a fundamental
understanding of scaling parameters.
A small subset of microreactor studies has been dedicated to reactions involving
solids, with different methods and strategies developed to prevent clogging. In most
cases, these are applications of flow over or past stationary reactive solids (solidsupported catalyst as a packed bed184-188 or a surface coating189,
190
), which prevents
clogging by immobilizing the solid phase. Such systems have the limitation of the
necessity to regenerate or recharge the catalyst, which may be difficult to remove in some
cases.
Microreactors have been applied to the formation of nanoparticles such as colloids191193
or quantum dots,63, 156 which remain suspended in solvent and thus flowable, although
these can sometimes lead to particle accumulation on surface, leading to clogging over
long time periods. In the cases where the solids in question are less easy to flow, they are
typically encapsulated in a separate liquid phase, either as droplets194-197 or in annular
138
flow,198 thus preventing their interaction with channel walls, and low concentrations and
amounts are produced to ensure flowability. However, contamination by oxygen, water,
or presence of certain solvents can influence the reactivity of many organic reactions.
Few works have discussed the precipitation of reactants199 or products in flow,200 and to
our knowledge, there has been no systematic study of solid formation and clogging in
microchannels.
In certain cases, particles in flow, such as cells or non-interacting particulates, can be
segregated and sorted using different hydrodynamic techniques such as flow focusing and
filtration.201-204
Certain of these methods, however, can limit reaction screening
applications because of the frequent need to remove and replace or clean filters.
Additionally, these techniques are challenged by high particle concentrations or by
particles that have electronic or electrostatic affinity for each other and for the flow path
surfaces, thus limiting their applicability to only a small number of organic systems. An
electrical potential may also be applied to sort particles in microfluidic systems.205,
206
Another approach to particle sorting is the application of acoustic standing waves to order
particles, such as the acoustic streaming of blood cells.207-210 More recently, we have
demonstrated the potential applicability of ultrasound to disrupt solids agglomeration and
enable chemical synthesis of a solids-generating reaction in flow,211, 212 while others have
applied ultrasonication to multiphase flow to enable polymerization in a microfluidic
system.200
The strategy for solids handling depends on the particular chemical
transformation; therefore, to provide the basis for a robust system of general applicability,
a deeper understanding of the clogging mechanism is necessary. This would allow for
the design of an important tool for synthetic chemical discovery and evaluation.
6.2
Palladium-catalyzed Amination
Many fine chemical synthesis routes require the formation of C-C and/or C-N bonds.
One such family of couplings occurs between an aryl halide compound and another
substituted aryl reagent, typically generating an inorganic salt as a byproduct. Thus, salt
formation is very common in such processes.146 A particularly useful organic synthesis
step for the formation of C-N bonds is the palladium-catalyzed amination, which has
become an important tool in organic chemistry, applied in the production of active
139
pharmaceutical ingredients, natural product syntheses, and fine chemicals. This reaction
has a relatively short residence time, high yields, and can be performed at fairly mild
conditions; however, it also forms inorganic salts due to the coupling of an aryl halide
with an aryl or aliphatic amine. In addition to aryl halides, aryl triflates and nonaflates
can be used as coupling partners to extend the scope of the reaction to a wider range of
starting materials.213-215 Nevertheless, many of the formed salts are insoluble in the
reaction solvents capable of performing C-N bond forming reactions. For this reason,
these reactions have been typically limited to traditional batchwise synthesis.
The particular chemistry selected as the example is the formation of biaryl amine 34
via the coupling of the aryl halide 4-chloroanisole (CA) 32 with the aryl amine aniline
(AN) 33 (Scheme 6.1), catalyzed by palladium coordinated to XPhos (2dicyclohexylphosphino-2',4',6'-triisopropylbiphenyl),216 a dialkylbiaryl phosphine ligand
with good stability in air, making it particularly easy to handle.217 The reaction requires a
fairly strong base, with sodium tert-butoxide (NaOtBu) selected based on its relatively
high solubility (~ 1 M) in the solvent of choice, 1,4-dioxane.
H2N
Cl
XPhos/Pd(OAc)2
+
H
N
NaOtBu
MeO
1,4-dioxane
34
o
32
33
80 C
Scheme 6.1. Palladium-catalyzed amination of 4-chloroanisole with aniline.
+ NaCl
MeO
It can be seen in Scheme 6.1 that one mole of sodium chloride is formed for every
mole of the desired product 34. Inorganic salts such as NaCl are generally insoluble in
1,4-dioxane, as well as in other solvents that may be used to carry out this reaction (e.g.,
toluene, tetrahydrofuran). In typical batch conditions, reactions of this type are typically
performed at reagent concentrations in the range of 0.5 to 1.0 M, producing rather large
amounts of solids. Moreover, at these concentrations, this reaction is complete within
several minutes, requiring short residence times and thus fast flow rates if performed in
flow. Thus, this chemistry is particularly applicable as a challenging model to study the
formation of solids in microfluidic systems.
140
6.3
Aminolysis Experimental Procedure
All reagents used for the aminolysis coupling were reagent-grade. Anhydrous 1,4dioxane was used as the solvent, and care was taken to ensure no contamination with
moisture. Two solutions in 1,4-dioxane were prepared for each experiment: one of
NaOtBu, typically 1.0 M, and one combining the remaining reagents (CA 32, AN 33,
XPhos, and Pd(OAc)2, typically 1.0 M, 1.2 equiv., 2 mol% and 1 mol%, respectively).
The base solution was always filtered through 0.2-μm Teflon-based inline Whatman
filters to remove potentially undissolved NaOtBu and any impurities (typically, NaOH,
which is not soluble in 1,4-dioxane). If unfiltered, the base solution alone, with no other
reagents present, was able to clog microreactor channels, often at the reactor inlet.
Figure 6.1. Experimental microfluidic system setup for the solids-generating coupling reaction. P, T,
and PID represent provided pressure and temperature and PID control, respectively. Dashed lines
indicate monitoring or electronic connection.
The flow of reagents was accomplished in a manner similar to that used in Chapter 3
and Chapter 4, using identical syringe pumps and Upchurch fittings.
Figure 6.1
illustrates the configuration of the setup applied in the solids-handling study.
The
reagents were delivered via 10-mL SGE or Hamilton Gastight glass syringes by a
141
Harvard Apparatus PHD2000 syringe pump. Pressure was continuously monitored by a
Honeywell stainless-steel pressure transducer (19C100PG4K), which was connected
directly into the primary reagent line (containing catalyst and the two coupling reagents)
upstream of the mixing of the reactants. The pressure transducer was excited using a 10V power supply, and the output was recorded with a National Instruments USB 9219 data
acquisition card connected to a Windows XP laptop computer running National
Instruments LabVIEW 8.5.1.
Upon exiting the reactor, the mixture passed through approximately 120 μL of 1/16”
FEP tubing (1000 μm I.D.) at ambient temperature before being collected in vial
continuously purged with argon at 1.7 psig. All other fluid connections were made using
¼”-28 PTFE fittings and 500 μm I.D. FEP tubing (IDEX Corporation). Images were
captured with a high-speed color CCD camera (JAI CV-S3200 series) connected directly
to a Windows XP computer.
At the start of every experiment session, pure solvent was flowed into both inlets at a
flow rate equal to that to be used in the experiment, and the pressure was recorded and
used as the baseline pressure drop ΔP0. When an experiment was terminated due to a
clog, the reactor was unclogged by applying pure solvent at increased pressure until the
blockade was forced through the reactor. This was followed by manual application of
several milliliters of water (in which the solids were soluble) at high flow rates to remove
any deposit on walls and of several milliliters of solvent to remove traces of water prior
to performing subsequent experiments.
6.4
Microreactor Design
6.4.1 Initial Microreactor Evaluation
Microreactor Setup
Initial microreactor experiments were performed using a microreactor design
previously developed for quantum dot particle synthesis.63 The microreactor design is
similar to, and served as the basis for, the device described in detail in Chapter 4. The
design contains a 120-μL reaction zone consisting of a single channel, 400 × 400 μm in
cross-section, comprised of 40-mm lengths connected by 180º turns with 400-μm average
142
turning radius. The inlet layout, mixing zone, and the halo through-etch between the
cooled port/mixing zone and heated main channel are identical to those of the reactor
described in section 4.3.1. Section 4.3.3 describes the heat transfer evaluation for the
thermal separation and for the fluid entering the reaction zone.
The microreactor was fabricated from 650-μm-thick silicon and Pyrex wafers using
the standard method described in section 4.3.1, using silicon nitride as the protection
layer. A compression chuck identical to the one described in section 4.3.2 was machined
out of 316 stainless steel, and Kalrez® o-rings were used for compression. The chuck
was cooled to 20ºC using a Thermo Scientific NESLAB RTE-7 refrigerating bath. The
temperature in the heated part of the microchannel was controlled by a Kapton flexible
heater (KHLV series, 28V) integrated with an Omega CN9000 series PID controller. A
packaged serpentine microreactor is shown in Figure 6.2.
Figure 6.2. Serpentine microreactor used for the initial solids handling tests, compression-packaged
and attached to a Kapton tape heater.
Clogging mechanisms
Initial investigations were performed to determine the primary mechanisms of
channel clogging and their correlation to the reaction conditions. The coupling chemistry
was performed in the microreactor at low concentrations, with 0.1 M of CA 32 as the
basis, at a reaction temperature of 80.0ºC. Clogging was observed in all cases within 10
residence times; however, the mechanism of the clogging was strongly dependent on the
flow rate.
143
At a flow rate of 20 μL/min, resulting in a residence time of 6 min in the heated zone,
large amounts of white solids began to form and agglomerate. These agglomerates were
able to flow fairly well along the channel straights; however, they became detained at the
sharp 180º turns, as shown in Figure 6.3a. Upon higher magnification, the agglomerates
appeared to be in the range of 100-300 μm (Figure 6.3b), or of the same order as the
channel width. The dimensionless pressure drop across the microreactor ΔP/ΔP0 rapidly
increased after about 5 residence times, leading to clogging to the point of no flow within
an additional 3 residence times (Figure 6.3c).
(b)
(a)
(c)
(d)
Figure 6.3. Microchannel clogging by solids agglomeration: (a) photograph of the microreactor
showing accumulation solids, primarily at 180º turns, with (b) a close-up of solids in the
microchannel; (c) dimensionless pressure drop across the microreactor during the experiment; (d) a
cartoon of flow-induced bridging between two flat plates.
For flow of particles that are only 3-4 times smaller than the channel dimension, it
has been shown that bridging takes place in micron-sized pore throats218-220 and
fabricated geometries,221 as illustrated in Figure 6.3d. However, this aspect ratio only
applies to stable particle suspensions that do not exhibit attractive interactions or change
144
size with time. In the case of our reaction, though, particles can grow due to the reaction
proceeding or can further agglomerate due to their ionic crystal interactions.
Additionally, particle-to-wall interactions in laminar flow can be caused by fluid flow
around turns, either because particles are forced to change their velocity or because their
momentum forces them off the streamline.222 This is consistent with our observation that
particle agglomerates tend to be concentrated around the sharp turns in the channel.
(b)
(a)
(c)
Figure 6.4. Microchannel clogging by wall deposition: (a) photograph of the microreactor showing
color change with surface coating; (b) dimensionless pressure drop across the microreactor during
the experiment; (c) a cartoon of flow constriction due to deposition between two flat plates.
Performing an identical experiment but with the flow rate increased to 70 μL/min (i.e.,
τ = 1.7 min), no large clusters of solids were observed, presumably because at this
concentration, the 1.7-min residence time was insufficient to produce a high yield.
However, the channels were observed to have changed in color, indicating a coating of
the surface (Figure 6.4a). The channels closest to the beginning of the reactor displayed
the greatest color change, indicating the fastest deposition. In this case, clogging was still
observed within a similar number of residence times as in the previous experiment
(Figure 6.4b).
Both deposition222-224 and nucleation followed by growth can lead to channel
constriction, as illustrated in Figure 6.4c. As the width of the channel decreases with
145
time, this will eventually lead to the small particles suspended in the bulk being able to
bridge the channel, leading to flow stoppage.
Assuming that the deposition rate on the walls of a channel is constant at any given
point along the channel length, the change in the channel dimensions with time can be
expressed as:
da db
=
= −α
dt dt
6.1
where a and b are the channel width and depth, and α is twice the wall deposition rate.
Therefore, the equivalent diameter for a square channel70, 71 will also change by the same
factor –α with time t. Thus, for laminar flow in a square channel, using the HagenPoiseuille equation, the pressure drop change with time will vary as follows:
DE4 ,0
ΔP
1
=
=
4
4
ΔP0 ( DE (t ) )
⎛
α ⎞
t ⎟⎟
⎜⎜ 1 −
⎝ DE ,0 ⎠
6.2
where DE is the channel equivalent diameter. Therefore, in the cases where the pressure
is changing only due to surface deposition, the change in pressure drop will follow a 4thorder increase with time, which matches well with the observed pressure drop trend in
Figure 6.4b.
6.4.2 Spiral-channel Microreactor Designs
Design for standard reagent introduction
To minimize the observed particle accumulation around the sharp turns, a
microreactor was designed to only have gradual, winding turns, as shown in Figure 6.5
and as previously discussed in Chapter 4. Detailed process run sheets and mask layouts
for all of the microreactors discussed in this section are given in Appendix A. The
reactor design was identical in functionality to the previously described serpentine
microreactor, retaining the same layout of ports and mixing zone, an equal volume, and
an identical thermal isolation of the reaction zone from the ports, allowing the addition
and mixing of reagents under cooled conditions (preventing reaction and solids formation
in the mixing zone) while the reaction section remained at temperature (e.g., 80.0ºC).
However, the nested spiral layout of the reactive channel provided a gradual change in
146
turning radius, with the sharpest turn having a radius of 4.4 mm rather than 0.4 mm for
the hairpin turns of the serpentine layout. Upon injecting reagents into this device at the
conditions of Figure 6.3 (i.e., τ = 6 min), we observed the flow of solids without the
accumulation shown in Figure 6.3b for several reactor volumes, with the solid aggregates
continuing to flow through the channel. Nevertheless, the microreactor wall (from top
down) gradually changed from transparent to white due to a coating of salts, as shown in
Figure 6.6. Pressure increased gradually, as in Figure 6.4b, and clogging eventually took
place.
(a)
(b)
cm
Figure 6.5. Silicon microreactor with a gradual spiral main channel to imitate a long straight path:
(a) illustration (showing the inlets and mixing area in green, main channel and outlet in black, and
halo etch for thermal isolation in violet) and (b) photograph.
(a)
(b)
Figure 6.6. Wall deposition in a spiral microreactor: (a) after 10 min (1.7τ); (b) after 45 min (7.5τ);
the rightmost straight channel is the outlet, and the adjacent channel is the inlet; the first curved
channel is closest to the outlet; the second is closest to the inlet.
In addition to the deposition on the walls, clogging was periodically observed as the
flow attempted to exit the reactor. Because the outlet is a through-hole etched into the
147
chip, the flow must necessarily take a sharp right-angle turn to exit the reactor. This
emulates the sharp turns seen in the serpentine microreactor and is an additional locus for
solids accumulation and clogging. In an attempt to address this concern, as well as to
expand the utility of this microreactor design, several design modifications have been
made to the spiral microreactor. First, to allow for longer operation before the onset of
clogging, as well as to provide additional space for any potential surface modification
treatments, the reactor channels were enlarged to be 500 × 500 μm in cross-section. To
accommodate this enlargement, 1-mm-thick silicon wafers were used for the
microfabrication. The same chip footprint and inter-channel distance were maintained,
thus yielding a reactive zone volume of 220 μm.
Figure 6.7. Redesigned spiral microreactor with enlarged channels and addition of a quench
(showing the inlets and mixing area in green, main channel in black, the quench and the outlet in
blue, and halo etch for thermal isolation in violet).
The most significant modification of the design was the introduction of a quench, as
shown in Figure 6.7, to terminate the reaction and to constrain the solids from contacting
the side walls of the channels. To that end, the quench was designed to be bifurcated to
surround the main reactive stream. Additionally, the quench is placed in the heated zone
to provide some residence volume for quenching, as well as a higher temperature for
better dissolution if a proper quench solvent is applied.
148
It was found that when water was used as the quenchant, rather than improving solids
exiting the reactor, it exacerbated the clogging situation. This was due to the water
contact causing the palladium catalyst to precipitate; in addition, some of the 1,4-dioxane
partitioned into the water, increasing the concentration of NaOtBu in the organic phase,
causing the base to precipitate, as well. Moreover, the residence time provided for the
quench was insufficient to effect dissolution of NaCl salt in the water phase. Thus, the
quench caused more solids to be generated than dissolved. However, a modicum of
success was found when only a low flow rate of 1,4-dioxane was flowed into the quench
stream. This was found to keep the solids-containing stream away from the side walls by
virtue of laminar flow, staving off clogging by several residence time intervals as
compared to no quench.
Sequential addition design
It was observed when performing this chemistry it the microreactor that the majority
of the solids are produced in the early section of the reactor channel, with smaller
amounts of solids produced with additional residence time. This is to be expected, as the
initial concentrations of reagents are the highest, and reaction rates typically decrease
significantly along the reaction length as reagents are consumed. To ameliorate the
tendency of the microreactor system to clog, it was proposed to add one or more reactants
sequentially along the length of the reactor, thus distributing its concentration and
resulting in a more evenly distributed production of solids throughout the reactor. In this
manner, nearly the same yield may be accomplished with less total solids within the
overall reactor volume.
To determine how the sequential addition of any particular reactor affects the overall
yield and formation of solids, the rate law of the model chemistry was necessary. Based
on the most recent reaction mechanism proposed for the organometallic-catalyzed
coupling of aryl halides with aryl amines225 and on the described mechanism of XPhos in
precatalyst and catalyst complexes,226 the mechanism for the model reaction was
proposed, as shown in Scheme 6.2. Mechanistic work had shown that the reaction with
base is the rate-limiting step and that the amine insertion may be reversible; thus, the
following rate law is derived:
149
Scheme 6.2. Proposed mechanism for the model chemistry.
d [Prod] k3 k4 [amine][base][Pd]tot
=
dt
k−3 + k4 [base]
6.3
This shows that the aryl halide does not participate in the reaction rate, while other
compounds are important to it. Additionally, if reaction step 4 is rate-limiting, k-3 may be
much greater than k4, making the reaction appear to be simply first-order in each of amine
and base at constant catalyst concentration, with a single observed rate constant.
This reaction was shown to be flowable in FEP tubing at very low concentrations,
where only small amounts of solids are generated. Based on yield vs. residence time data
obtained in such experiments, the observed overall reaction rate constant was fitted.
Using said reaction rate constant, the effects of sequential addition (either in 10 or 100
additions) of either catalyst or the aryl bromide are shown in Figure 6.8 and compared to
standard single addition.
For the calculation, it is assumed that the species to be
sequentially added is evenly distributed between the specified number of inlets that are
equally spaced along the reactor length, with the first inlet occurring at the beginning of
the heated reaction zone. The conditions are assumed to be equivalent to those used in
Figure 6.3 (0.2 M aryl bromide in syringe, 10 μL/min flow of each of the two syringes).
Figure 6.8 shows the amount of product flow along the reactor (given in reactor volume),
150
and the final conversion is reported in the table on each graph. The total amount of solids
in the reactor at steady state is given by the integral of each curve and is reported on the
graph as “total solids,” in arbitrary units of μL×μmol/min. It can be seen that there is
little gain from 100 sequential additions as compared to 10. However, splitting the amine
into 10 smaller infusions reduces the total solids in the reactor by 43%, while the yield
only decreases by 13% with no residence time change. A smaller but similar effect is
observed by splitting the catalyst.
2
(a)
1.6
Product flow rate (μmol/min)
Product flow rate (μmol/min)
2
1.8
1.4
1.2
1
0.8
conv:
0.6
total solids
10 injections
94.1%
90.9%
100 injections
Straight addition 90.0%
0.4
0.2
164
134
126
(b)
1.8
1.6
1.4
1.2
1
0.8
conv:
0.6
0.2
0
0
0
20
40
60
80
100
Volume of the reactor (microliters)
120
total solids
10 injections
94.1%
81.0%
100 injections
Straight addition 78.5%
0.4
0
20
40
60
80
164
95
86
100
120
Volume of the reactor (microliters)
Figure 6.8. Sequential addition of (a) catalyst and (b) aryl amine into the C-N coupling reaction.
It was decided to modify the initial spiral reactor with the 400 × 400 μm, 120 μL
reaction zone to provide sequential addition of one reagent stream in ten equally spacedapart locations of the second stream, with a quench following the termination of the
reaction zone. The sequential addition channel was designed as a bifurcation of the
initial stream, with the two channels being threaded between the main flow channel, one
flowing parallel to the main stream starting from the inlet, and the other flowing counter
to the main stream starting from the outlet. To determine the necessary lengths and
widths of each sequential addition sub-channel to provide equal flow to each of the ten
additions, the Hagen-Poiseuille model for rectangular channels was used,70,
71
as
discussed in Chapter 2 and Chapter 3:
ΔP =
128μ De4 L
π
Q = QR
6.4
where R is the combined term, representing resistance, equivalent to a circuit diagram
(with flow rate standing in for current and pressure drop for potential). The equivalent
diameter De is given as:
151
128ab3
D =
πK
4
e
6.5
where a and b are the large and small sides of the rectangular cross-section, respectively,
and K is an empirically obtained function of the ratio of a to b and can be modeled as a
power law. Using the aforementioned electrical circuit analogy, a resistor circuit was set
up for the sequential addition from a single source being split into first two and then five
each, adding along ten equally spaced locations along the main channel (Figure 6.9).
1
/2Qs
Qin + Qs
Qin
Qs
P12
P1
/2Qs
1
Rs1
/10Qs
Rin
Ri1
Rsc
P14
Ri8
P11
P17
1
Rc
Rc
Rsc
/10Qs
Ri7
Rsc
P18
P3
Ri2
Rc
Ri6
P9
P19
Ri3
Rc
Rc Rsc
Rsc
P5
P16
Ri4
Ri5
P8
P20
1
/10Qs
Rc Rsc2
Rc
Rc
Rsc1
P6
Rc
P10
Rsc
P4
P15
Rout
Rs2
P2
P13
P=0
1
P7
1
/10Qs
Figure 6.9. Sequential addition reactor design as a circuit diagram; main channel is highlighted in
blue, and addition channel is highlighted in green.
Several assumptions were made to enable the solution of the circuit diagram. First,
the design was made to function with equal flow rates of the two reagent streams.
Second, the viscosities of the reagent streams were assumed to be constant. Third, there
was assumed to be no change in volume, density, or viscosity with mixing, and the
change in viscosity with produced solids was neglected.
152
Solving the circuit diagram allowed the design of the sequential reactor, shown in
Figure 6.10. The sequentially-added-reagent channel was to be 93.2 μm wide and etched
to the same depth as the main channel, 400 μm. The connections made between that
channel and the main one were to provide a sufficiently high pressure drop as to prevent
any possible backflow into those channels.227 Thus, the additions #1-4 and #7-10 were
designed for a depth of only 10 μm. Additions #5 and #6 (the terminal additions of the
two channels following the bifurcation) were 400 μm deep, but with the channels
narrowing following the penultimate additions to 50.0 and 55.2 μm in width,
respectively. To ensure equal flow between the two main addition channels, the channel
providing additions #1-5 was made 62.5 mm longer than was required by layout
geometry. The respective widths of the shallow-etch inlets #1-4 were 41.4 μm, 54.6 μm,
72.5 μm, and 92.0 μm, and for inlets #7-10, 102.8 μm, 71.0 μm, 50.3 μm, and 37.5 μm.
Figure 6.10. Sequential addition spiral microreactor, showing the inlets, the quench, and the outlet
in blue, the main channel in black, the sequential addition main channels in green, the shallow-etch
sequential addition inlets in orange, and the halo etch in violet.
The shallow inlets were fabricated by an addition of a nested mask to the fabrication
process, in a method identical to the etching of the manifolds and mixing cavity of the
first-generation micromixer, described in section 2.3.1. This required the additional steps
of virgin wafer oxidation and of BOE etch during feature development. These mask
153
features for these inlets were printed to be 1 μm wider than the aforementioned values,
and the inlets were etched to 10.5 μm, in both cases to account for the growth of 500-nm
oxide layer as the final step before wafer bonding.
The flow distribution sensitivity to etch variations was found to be reasonable
considering the variation range. There was very little sensitivity to the depth of the main
etch for a variation of up to 30 μm across the chip or off from the desired depth. For the
short etch of the shallow features, it was found that, with proper care, etching can be
stopped to within 0.3 μm of the desired depth, with an across-the-chip variation of no
more than 2%. This level of variation was calculated to be able to maintain the desired
flow rates through each inlet to within 15%.
While not ideal, this nevertheless
accomplishes the desired target of sequential addition along the reactor volume.
The inlets are located such that a pulse sent into the sequential addition channel,
despite being split into ten smaller pulses, would nevertheless recombine within the main
channel. Thus, attempting a pulse RTD may show some limited evidence of channeling
but would not provide information on whether all inlets are operational. To obtain this
desired information, flow visualization techniques were undertaken.
To visualize the sequential addition, several methods were applied. First, toluene was
flowed into the sequential addition channels as water was flowed into the main channel,
both at flow rates of 20 μL/min. The formation of fine droplets (which rapidly coalesced
into round slugs of ~300 μm in diameter) was observed in all ten locations of the
sequential addition inlets. Inlets #5 and 6 seemed to produce more droplets than any of
the other ones. However, that is to be expected and is not indicative of flow of miscible
fluids, as there is a large surface tension between water and toluene, which changes the
pressure drops of the inlets, more strongly favoring the ones with wider apertures.
As a second visualization method, a flow of Rhodamine B solution in water was
added to the main stream of pure water at rates of 20 μL/min each; UV illumination was
used to fluoresce the dye, recording the visuals with the CCD camera. During the initial
transient, fluorescing dye was seen entering the main channel in several of the inlets,
although fluorescent flow rapidly overtook the channel, making inlets difficult to
observe. However, at steady state, it can be clearly seen in Figure 6.11 that the brightness
of the channel, proportional to the concentration of Rhodamine B in the flow, is gradually
154
increasing along the reactor length, confirming the gradual distribution of the
concentration of the “reagent.”
Figure 6.11. Sequential addition spiral microreactor with a solution of Rhodamine B sequentially
added to the main flow of pure water.
Applying this reactor to the C-N coupling reaction at the conditions used in Figure
6.3 met with mixed success. While it was clearly observable that the flow of solids was
much more distributed throughout the reactor and that less total solids was flowing, the
reactor nevertheless clogged within 10 residence times, primarily at the outlet.
Additionally, wall deposition was observed during the experiments, as well. Therefore,
this confirms that the quench design is inadequate to allow the solids to continuously
flow through the sharp 90º turn when exiting the reactor. Moreover, other methods are
required to prevent or assuage wall deposition.
6.5
Fluoropolymer Surface Modification
Experiments using the studied coupling chemistry in simple fluorinated ethylene
propylene (FEP) capillaries of 500 or 1000 μm as model reactors demonstrated them to
be less prone to clogging due to surface deposition. Thus, one possible approach to
155
minimizing wall deposition or growth is the modification of microreactor surfaces with a
fluorous chemistry, combining the benefits of the silicon microdevices with fluorous
surfaces. To that end, a silicon microreactor with 500 × 500 μm channels and a quench,
described in the previous section, was coated with polytetrafluoroethylene (PTFE).
Mixing zone
Main channel
Water/hexane
Silicon oxide
PTFE
Figure 6.12. Silicon microreactor before (top row) and after (bottom row) surface coating with
PTFE; from left to right: mixing zone, reaction zone, and biphasic flow of aqueous fluorescein and
hexane.
The reactor surface was first pretreated with a solution of 1H,1H,2H,2Hperfluorodecyltrichlorosilane (from Alfa Aesar) in hexane (30 μL in 15 mL) in a
moisture-controlled glove box. An aqueous suspension of nanoparticles of PTFE (TE3859, Tg = 337ºC), obtained from DuPont, was flowed into the reactor by annular gasliquid flow. Industrial-grade nitrogen gas (Airgas) was regulated to the desired pressure
for annular flow. Images of the microchannels were captured with a Canon PowerShot
S5IS camera fitted on a Diagnostic Instruments Leica MZ12 microscope. Hexane and
fluorescein dissolved in water were used to demonstrate the surface wetting properties.
Figure 6.12 shows that the coating of silicon microreactors with PTFE switches the
wetting characteristics of the channel from the hydrophilic of silicon oxide to the
hydrophobic of PTFE. Although a fairly even and uniform layer of PTFE is formed on
the channel surfaces, cracks and surface imperfections nevertheless present nucleation
points for particle deposition and for attack by the strong organic and inorganic bases
routinely used in C-N bond formation reactions.
156
An attempt was made to improve the adhesion of the PTFE layer to the reactor surface
by preliminary surface modification using the DRIE passivation step (see section 2.1.1).
Following the completion of the DRIE etch, a 6-min passivation step was performed,
depositing ~0.6 μm of amorphous fluoropolymer. Figure 6.13 shows an SEM image of
the coated channel surface following all subsequent cleaning steps and immediately
preceding the bonding step. It can be seen that the polymer surface contains numerous
cracks. However, it was hoped that the coating of PTFE would be enhanced by the
presence of a chemically similar fluorous compound that has been deposited onto silicon
under plasma conditions.
Figure 6.13. DRIE fluoropolymer passivation coating in a microchannel.
Several thusly prepared devices were coated with 2 or 4 layers of PTFE to produce a
robust film. An aqueous solution of 20 wt% KOH was then flowed with the reactor at
60ºC, intending to rapidly reveal any imperfections in the coating, as the base would
begin to rapidly and visibly etch the unprotected silicon beneath the PTFE film. Indeed,
this was seen to occur within approximately 10 to 20 minutes of the beginning of the test.
Following the test, optical microscopy observations of the channels revealed that the
PTFE film did in fact delaminate from the channel walls, allowing wall etching to occur
(Figure 6.14a).
Additionally, in some locations, poor silicon-Pyrex bonding was
revealed, as evidenced by etching reaching far past the channel walls (Figure 6.14b). The
157
latter is believed to have been caused by agglomerates of the passivation fluoropolymer
that have remained on the top surface of the wafer, seen as white clusters in Figure 6.13.
Figure 6.14c shows that the PTFE coating at the inlets does not cover the 90º step
entering the reactor, thus allowing liquid to bypass the polymer film when flowing into
the reactor. As this has not occurred in the previous tests with water and hexane (as
evidenced by the lack of menisci in Figure 6.12), this phenomenon may be due to the
KOH solution being an aggressive etchant of silicon at these temperatures. Therefore,
while the surface modification may not provide adequate protection of the reactor
channel surface, it does hold promise as a method of switching the wetting characteristics
of microreactors under certain conditions.
(a)
(c)
(b)
Figure 6.14. PTFE-coated microchannel following flow of 20 wt% KOH at 60ºC; (a) delamination of
PTFE film and wall-etching; (b) etching extends into unbonded vesicle; (c) PTFE film inlet.
6.6
Application of Acoustics
6.6.1 Acoustic Irradiation
It is commonly known that sound waves can impose a force on a system of
particles.228 To determine whether such a force can prevent particles from agglomerating
on channel walls or with each other, we investigated the influence of acoustic waves on
microreactor clogging during palladium-catalyzed C-N bond forming reactions. The
primary force exerted by an acoustic standing wave in the radial direction on a particle
flowing in a microchannel, Fr, has previously been described as:207, 208
⎛ 2π 2 p 02 r 3 β S
Fr = −⎜⎜
3λ
⎝
158
⎞
4πx ⎞
⎟ ⋅ φ ⋅ sin ⎛⎜
⎟
⎟
⎝ λ ⎠
⎠
6.6
where p0 is the acoustic pressure amplitude, r is the particle radius, βS is the
compressibility of the solvent, λ is the wavelength of the standing wave, x is the location
along the wavelength, and φ is a contrast factor, described as:207, 208
⎛ 5ρ − 2 ρ S
φ = ⎜⎜ P
⎝ 2ρ P + ρ S
⎞ βP
⎟⎟ −
⎠ βS
6.7
where ρ is density, and the subscripts P and S denote the particle and solvent,
respectively. One observes in equation 5.6 that the primary force is strongly dependent
on particle radius; thus, the acoustic force weakens as the particle diameter is reduced.
Furthermore, the primary force is driven by differences in density and compressibility,
described by the contrast factor. The acoustic pressure amplitude also influences the
primary radiation force and is dependent on the voltage and frequency under which
piezoceramic operation takes place.
Such forces have been exploited to order and
separate particulates and biological matter when frequencies are in the range of MHz.207,
208, 229, 230
We wished to determine whether a standard ultrasonicating bath (VWR 50HT)
would have similar effects and whether it would be capable of disrupting plugging by
solids.
The waveform produced by the transducer of the ultrasonic bath was recorded with a
National Instruments USB 9219 data acquisition card using National Instruments
LabVIEW 8.5.1 software by measuring the voltage supplied to the transducer. The
waveform was observed to have several different periodicities, as shown in Figure 6.15ad. The sinusoidal waves had a maximum voltage ranging from 25 to 500 V.
These figures were used to determine the frequencies of the acoustic signal and to
create a function capable of reproducing it. The acoustic modes were seen to be a
sinusoidal mode at 41.5 kHz (Figure 6.15d), a dual-sinusoid (every second peak is at
half-height) at an overall frequency of 1 kHz, a “ringing” mode at 120 Hz, and a ramp-up
followed by a drop at 2.95 Hz. The first mode can be easily written as:
f1 (t ) = sin(2π ⋅ 41.5 kHz ⋅ t ) = sin(260752t )
6.8
The second mode, a dual sinusoid, is a sum of a sine at twice the frequency and a
half-value cosine:
1
1
f 2 (t ) = sin(2π ⋅ 2000 Hz ⋅ t ) + cos(2π ⋅1000 Hz ⋅ t ) = sin(12566t ) + cos(6283t ) 6.9
2
2
159
50
600
(a)
400
(b)
f = 120 Hz
40
f = 2.95 Hz
30
20
Voltage
Voltage
200
0
-200
10
0
-10
-20
-30
-400
-40
-50
0.28
-600
0
0.2
0.4
0.6
0.8
0.285
100
(c)
0.295
0.3
t (sec)
t (sec)
80
0.29
200
f = 1 kHz
(d)
f = 41.5 kHz
150
60
100
20
Voltage
Voltage
40
0
-20
50
0
-50
-40
-100
-60
-150
-80
-100
0.324
0.326
0.328
0.33
-200
0.0502
0.05022
t (sec)
0.05024
0.05026
0.05028
0.0503
t (sec)
Figure 6.15. Ultrasonication bath waveform at different data capture rates and resolutions.
The ringing mode is caused by a second sine wave added to the one in eq. 5.8, this one
with a frequency such that the difference in frequencies of the two sine waves is the
frequency of the ringing:
f 2 (t ) = sin(2π ⋅ (2000 Hz+120Hz) ⋅ t ) = sin(13320t )
6.10
The ramp was smoothed and fitted to determine the best approximation function, as
shown in Figure 6.16. The exponential is a ramp with a period of 0.3386 seconds; thus, it
can be represented by a Laplacian:
f P (t ) = e9.5t ( u (t ) − u (t − 0.3386) )
6.11
where the subscript P represents a single period, and u(t) is the Heaviside function. Thus,
taking the Laplace of this yields:
160
L { f P (t )} = L {e9.5t ( u (t ) − u (t − 0.3386) )} = L {e9.5t ( u (t ) )} − L {e9.5(t −0.3386) +3.2167 ( u (t − 0.3386) )} =
=
1
e3.2167 e −0.3386 s 1 − e3.2167 −0.3386 s
−
=
s − 9.5
s − 9.5
s − 9.5
6.12
and the Laplace of the periodic with a period of 0.3386 seconds is therefore
L { f (t )} =
1 − e3.2167 −0.3386 s
( s − 9.5) (1 − e−0.3386 s )
6.13
Finally, multiplying the inverse Laplace of this function by the sum of equations 5.85.10 would yield an overall function with the correct range of voltage and the appropriate
periodicities:
350
300
Voltage
250
y = e9.5x
200
150
100
50
0
0
0.2
0.4
0.6
0.8
t (sec)
Figure 6.16. Smoothing of the absolute values of the data in Figure 6.15, with a fitted exponential.
⎧⎪ 1 − e3.2167 −0.3386 s ⎫⎪⎛
1
⎞
f (t ) = L−1 ⎨
sin(260752t ) + sin(13320t ) + sin(12566t ) + cos(6283t ) ⎟
−0.3386 s ⎬ ⎜
2
⎠
) ⎪⎭ ⎝
⎪⎩ ( s − 9.5) (1 − e
6.14
161
6.6.2 Integration of Acoustics with Silicon Microreactors
One target goal is to be able to deliver acoustic radiation to the silicon microreactor in
the most efficient manner possible. To that end, a chuck was designed to couple a
piezoceramic horn directly to an aluminum chuck typically used for heating the
microreactor (Figure 6.17). The chuck is based on the design of the heating chuck for the
four-port halo-etched microreactors described in section 5.3.2, accommodating two 35 W
cartridge heaters to provide temperature control, with an identical PID control setup. The
two modifications to the chuck include a rim to precisely position the silicon
microreactor and a threaded shaft protruding from the rear of the chuck. The rim serves
to act as a barrier preventing the chip from sliding off. The shaft is threaded to couple
directly with a piezoceramic stack identical to the one used in the aforementioned VWR
ultrasonicating bath.
The overall setup maintains full visibility of the reaction and
quench area of the silicon chip.
The chuck assembly includes the chuck base pictured in Figure 6.17a and an
aluminum frame, as shown in Figure 6.17b. The frame is lined with a Viton gasket that
provides compression to prevent reactor breakage during the activation of acoustics. The
Viton gasket is compressed against a 3/8”-thick glass piece that covers the reactor portion
resting in the chuck. The chuck base has four threaded screw holes for #6-32 screws,
which are then held in place with capture locknuts (McMaster 91831A007). The latter
are necessary to prevent the assembly from shaking loose during operation with
ultrasound.
(a)
(b)
Figure 6.17. Aluminum chuck for microreactor heating and direct acoustic radiation; (a) rendering
in SolidWorks (no threading is shown on the coupling stem); (b) photograph of a piezoceramic horn
coupled to the chuck, with the top frame attached.
162
To provide acoustic radiation, the previously used ultrasonic bath was disassembled,
and the internal piezoceramic horn was disconnected. The piezoceramic horn coupled to
the microreactor was then connected to the leads within the ultrasonicating bath, thus
ensuring that identical power and waveform generated by the bath are delivered to the
silicon device. This provides continuity and allows direct comparison with previous
research.
Because the piezoceramic horn is electrically live and the coupling chuck is made of
aluminum (and thus conductive), care must be taken during handling and operation of
this system. Additionally, a wire thermocouple cannot be used inside the chuck or
contacting the silicon because of the electrical disruption from the piezoceramic element.
To monitor the reactor temperature, a flat surface thermocouple (Omega® CO1-K) was
placed between the top surface of the reactor and the glass compression piece. Based on
the Comsol simulation presented in section 5.3.3, a difference of no more than 0.2ºC was
expected between the surface thermocouple and the temperature in the silicon channel
directly beneath it at a setpoint of 80ºC.
6.6.3 Effect of Acoustics on Reaction Parameters
Ultrasound has proven to be a useful tool in enhancing the rate of organic
transformations,231,
232
including reactions catalyzed by palladium.233 It is generally
believed that the energy emitted from cavitations can enhance reaction rates in batchscale systems that have temperature gradients. Recently, the research work of others has
elucidated that ultrasound enhances mixing in microfluidic systems.230,
234, 235
The
question thus developed of whether acoustics applied using an ultrasonicating bath or its
piezoceramic stack could create observable temperature and mixing effects that would
impact the palladium-catalyzed C-N bond formation reaction in microscale flow.
Effect of acoustics on reaction flow temperature
Previous work on applying acoustics to study the continuous-flow C-N coupling
shown in Scheme 6.1 was performed primarily in FEP capillary tubing (0.5-1.0 mm ID,
120 μL in volume) at conditions equivalent to ones described in section 6.3, using the
aforementioned ultrasonication bath and a PID-controlled immersion heater to obtain the
163
desired temperatures. The coupling reaction was carried out at 80.0ºC and a residence
time of 1 min and varying the catalyst loading from 0 to 1 mol%. Without the application
of ultrasound, the reaction yield was reduced at any given catalyst loading, as shown in
Figure 6.18, corresponding to approximately a 20% decrease in observed reaction rate.
These results suggest that the acoustic irradiation has an effect on the reaction rate, by
increasing the reaction temperature and/or reducing axial dispersion.
Figure 6.18. The influence of ultrasound on product yield for different catalyst loadings.
It is supposed that, although the cavitation-driven heating effect exists only on the
microscale, the energy thus delivered will dissipate to the fluid, raising its bulk
temperature by an observable degree. To investigate the effect of acoustic irradiation on
temperature in flow, a fine-gauge wire thermocouple (Omega® 5TC-TT-40-36) was
threaded into the FEP capillary reactor described in the previous section, which was
immersed into the ultrasonicating bath identically to the experiments.
Prior to the
beginning of the experiment, both the fine-gauge wire thermocouple and the one used for
monitoring and controlling the bath were placed adjacent to each other within the bath,
and the bath was heated to 80ºC. The readings of the two thermocouples coincided to
within 0.1ºC. With the fine-gauge wire thermocouple inside the FEP tube, water was
flowed into the tube reactor at 120 μL/min, and the bath was heated to 80ºC. Upon
reaching that temperature (which was read by both thermocouples), the acoustic
irradiation was switched on. At the instant of activation, the temperature both in the bath
and within the tubular reactor increased by 3ºC. However, it rapidly began to decrease
164
until the temperature in the bath reached 80ºC. While the temperature within the tube
responded more slowly, it also decreased to a steady state of 80ºC. This indicates that
while the acoustic irradiation does provide bulk observable thermal energy, the PID
control of the heating bath compensates for this, maintaining the setpoint bulk
temperature. Thus, in an FEP tube reactor in a heated ultrasonic bath, it is unlikely that
the acoustic irradiation contributes to reaction acceleration by thermal effects.
This experiment was also performed on the 220-μL silicon chip described in section
6.4.2, packaged in the aforementioned ultrasonication chuck.
The fine-gauge wire
thermocouple was compared to the surface thermocouple tied to the chip thermal control
setup, to satisfactory results (i.e., displaying identical temperatures). The fine-gauge wire
thermocouple was then threaded into the reactor through the outlet port up to the
intersection of the main reaction channel and the quench channels. To prevent the
thermocouple from being short-circuited by the electrical potential from the live
piezoceramic horn, the back and edges of the chip were covered with electrical tape prior
to packaging in the acoustic chuck. Based on finite element modeling (section 5.3.3), a
difference of < 0.2ºC was expected between the surface thermocouple and the threaded
wire one at a chip setpoint of 80ºC. This corresponded to the observed difference without
acoustic radiation, with water flow into the reactor of 60 μL/min through each of the two
inlets and the quench. Upon activating the acoustic radiation, the temperature inside the
reactor immediately increased by 3ºC. Interestingly, the temperature read by the surface
thermocouple increased by 1ºC. Both began to decrease after the initial spike, with the
surface temperature attaining a steady state of 80ºC (as it was controlled to that setpoint),
and the temperature inside the reactor attaining a steady state of 81.5ºC, slightly above
the setpoint. Therefore, inside a silicon device, where the temperature is monitored
outside a medium affected by cavitation, the temperature appears to be above the setpoint
by 1.5ºC, which may increase a typical catalytic reaction rate by 10-15%.
Effect of acoustics on residence time distribution
To study whether the effects of ultrasonication, such as cavitation, bubble formation
and collapse, and standing wave nodes, contribute to enhanced mixing, experiments were
performed in the FEP tubing setup identical to the one previously used for the aminolysis
165
reaction study. A length of 1 mm ID tubing with approximately ~350 μL in volume was
used, with 240 μL of it submerged in the ultrasonic bath. Fluid was delivered by a
syringe pump. Residence time distributions were measured using the setup discussed in
section 2.3.3, with a custom-made flow cell that aligned the optical fibers to allow the
light to pass directly through the FEP tube for collection. Water was used as the medium
fluid, and pure acetone was injected as a 5-μL pulse.
Evaluation was performed to compare the residence time distributions at room
temperature with and without acoustics at 120 μL/min flow. Each experimental condition
set was performed in triplicate, with very good reproducibility between data sets at the
same conditions.
Figure 6.19 shows representative RTD curves with and without
acoustics, showing no significant differences, as evidenced by the very similar τ and σ
values shown next to the corresponding curves.
The switching of the valve often
entrained a small bubble, resulting in the sharp spike seen in the RTD curves.
Next, we wished to determine whether a marked difference would exist in the
presence of solids. Silicon dioxide powder (Sigma-Aldrich, 0.5-10 μm, 80% between 1
and 5 μm) at 0.15 vol.% was introduced into both the water being flowed and the acetone
being injected. The flow was stopped and the syringe was agitated following every
experiment to ensure even dispersion.
Here, flow rates of 120 and 240 μL/min,
corresponding to 1 and 2 min residence times, were examined, also in triplicate. Good
reproducibility was also observed, although in the case of flowing solids with acoustics,
the number and location of bubbles seemed to vary stochastically. While the three curves
for that condition had similar profiles, the number and location of spikes (corresponding
to bubbles) varied. Representative RTDs with and without acoustics are shown in Figure
6.20 along with the τ and σ values of the corresponding curves.
In this case, no
significant difference was observed due to acoustics at 240 μL/min, where no bubbles
were entrained or formed. However, at 120 μL/min in the presence of solids, bubbles
were formed in the flow, which both reduced the residence time by 15% and significantly
decreased the variance (σ) from 54 to 29 s. It has been shown in the past that a gas-liquid
slug flow produces a much tighter RTD,236 resulting in plug-flow-like behavior of the
liquid. This outgassing, which is even more evident at the 80ºC reaction conditions,
would contribute to reduced dispersion, which, at 120 μL/min in tubing used, can result
166
in 3-5% higher observed reaction rates (as discussed in section 5.6). Combined with the
temperature effect, this may explain the phenomenon observed in Figure 6.18.
4
With Acoustics
No Acoustics
3.5
τ = 167 s, σ = 31 s
τ = 167 s, σ = 30 s
3
C(t)
2.5
2
1.5
1
0.5
0
0
100
200
300
Time (s)
Figure 6.19. Residence time distribution for liquid flow at 120 μL/min with and without ultrasound
irradiation.
4
2
With Acoustics τ = 177 s, σ = 29 s
τ = 209 s, σ = 54 s
No Acoustics
3.5
3
2.5
τ = 76 s, σ = 21 s
τ = 82 s, σ = 20 s
1
C(t)
C(t)
With Acoustics
No Acoustics
1.5
2
1.5
1
0.5
0
0.5
(a)
0
0
(b)
-0.5
100
200
300
400
0
50
Time (s)
100
150
200
Time (s)
Figure 6.20. Residence time distribution for solids-containing flow with and without ultrasound
irradiation at (a) 120 μL/min and (b) 240 μL/min.
6.6.4 Effect of Acoustics on Solids Handling
Particle size analysis
In the previous work on the C-N coupling chemistry in question (see section 6.6.2),
the outflow of the reaction was collected for each of the data points shown in Figure 6.18
and analyzed for particle size distribution to observe the effect of acoustics on flow. The
reaction mixture was collected under argon into vials of 1,4-dioxane (diluted by a factor
of 2) at room temperature, and the samples were analyzed using a Malvern Mastersizer
2000 laser diffraction analyzer, with the results shown in Figure 6.21. With acoustics,
167
particles ranged from approximately 0.15 to 36 μm in diameter, with the different catalyst
loadings (and thus different reaction conversions) resulting in identical relative particle
size distributions but different particle concentrations. This indicates that there is very
little particle growth with reaction; most newly formed salt seems to form new particles.
The collection of particles in the absence of acoustic radiation, however, revealed
particles in the range of 0.15 to 112 μm, as shown in Figure 6.21b. This observation
demonstrates that ultrasonic forces reduced the effective maximum particle size, which in
turn prevented bridging. The observed particle modes centering at 10-20 μm and 40-60
μm likely indicate particle agglomerates, with the latter being prevented or disrupted by
acoustic forces. Thus, the particle-to-particle interactions and hydrodynamic conditions
that lead to clogging can be overcome by applying external forces such as ultrasonic
irradiation.
(a)
(b)
Figure 6.21. Particle size distribution for the reaction samples in Figure 6.18 (a) without ultrasound
irradiation and (b) with ultrasound irradiation.
These results provide insight on reactor design for amination reactions to prevent
bridging. From Figure 6.21a, the aspect ratio (D/a) of the largest particle size in the
presence of ultrasound is 36 μm, while without ultrasound, this value is 115 μm. This
implies that microreactors with channels of 400 μm could potentially synthesize NaCl
particles in this C-N coupling reaction without clogging, as the aspect ratio D/a is 11 and
should thus be flowable.
While this neglects effects such as wall deposition and
interparticle attraction, this shows promise for the flowability of this reaction in a silicon
microreactor with application of acoustics.
168
Acoustic irradiation of silicon microreactor with sequential addition
To observe the ability to perform the solids-generating C-N coupling reaction in
microscale flow, several techniques to mitigate clogging have been combined in a single
experiment.
The spiral layout silicon microreactor featuring sequential addition
(described in section 6.4.2) was packaged into the ultrasonicating chuck (described in
section 6.6.2). The C-N coupling reaction was performed at the reagent concentrations
first described in Figure 6.3, based on 0.1 M of 4-chloroanisole. Because the base must
be kept separate from the reagents and the catalyst prior to reaction, as it activates the
catalyst, it was decided to add the base sequentially to the stream containing the
remaining components. As the reaction was in the past observed to be essentially firstorder in base, the effect was expected to be as predicted in Figure 6.8b.
20
Pressure Drop (ΔP/ΔP0)
18
16
14
Qi = 20 μL/min
12
Qi = 7.5 μL/min
10
8
10
reactor
volumes
6
4
8 reactor
volumes
2
0
0
1000
2000
3000
4000
5000
6000
Time (s)
Figure 6.22. Dimensionless pressure drop as a function of time for the C-N coupling in the acousticsirradiated sequential-addition silicon microreactor.
In this setup, a biphasic quench was used, consisting of water and butyl acetate,
delivered at equal flow rates of each of the other reagents via a tee to the quench port.
This mixture was selected to allow dissolution of solids (in water) while preventing the
precipitation of catalyst and base (by extraction to butyl acetate). Additionally, as there is
no mixing zone in this device, no cooling was used for the inlets and outlet, allowing the
reaction to be terminated by contact with water. Thus, the mixture with solids remained
169
heated till past the outlet, improving the solubility rate of the solids and helping prevent
agglomeration.
This reaction was performed at three different flow rates, with each reagent being
flowed at 60 μL/min, 20 μL/min, and 7.5 μL/min; corresponding average yields were
46%, 58%, and 63% (with conversions equal to yields). Figure 6.22 shows the pressure
data with time for the higher two flow rates in this experiment, normalized to the baseline
at the start of each experiment. At the highest flow rate, no clogging was observed for 10
residence times, at which point, the flow rate was decreased. At the second flow rate, the
pressure remained flat, with occasional small increases followed immediately by returns
to the baseline. After 10 residence times, the flow rate was further decreased. At the
lowest flow rate, a deposit on walls was observed in several places in the reactor.
However, no increase in pressure was observed for 9 residence times, after which, a
gradual but accelerating increase was observed, consistent with 4th-order dependence on
time, indicating constriction of the walls.
Overall, this is a highly promising result, indicating that at certain conditions, flow of
solids-generating reactions is possible in silicon microreactors. This may greatly expand
the range of reactions that can be screened and studied kinetically using microreactors as
tools. Additionally, this suggests that in the presence of acoustics, wall deposition does
not take place at any significant rate with sufficiently high flow rates (and thus high
shear), suggesting optimal an operating regime for solids-containing flows in
microchannels.
6.7
Conclusions
The preparation of a biaryl amine via a palladium-catalyzed amination represented a
case study for understanding how to handle salt byproducts in microsystems. Studying
this C-N coupling reaction in microreactors, we have found that the formed solids lead to
plugging of the microchannels predominantly via two mechanisms: bridging of particles
across the channel and constriction of walls due to a salt deposit.
Several approaches were investigated as means to alleviate the solids plugging issue.
By creating a reactor with the reaction zone containing only gradual, winding turns, it
was found that bridging can be minimized, as compared with serpentine channels.
170
Sequential addition of one reagent (that participates in the numerator of the reaction rate
law) can also decrease clogging by distributing the formation of the solids throughout the
reactor, rather than allowing the majority of the solids to be formed in the initial section.
The addition of a quench stream with a biphasic quench can help solubilize the solids
while retaining the organic components in solution. Moreover, with sequential mixing,
there was no need for cooling of the inlets, allowing the quenched stream to remain
heated as it exited the reactor, thus reducing clogging at the outlet.
The presence of acoustic irradiation reduced the effective particle sizes of salt
byproduct by disrupting the formation of large agglomerates, thus preventing bridging in
microreactors.
It is likely that the magnitude of the acoustic forces exceeded the
hydrodynamic conditions and particle-to-particle interactions that led to bridging.
Additionally, the cavitation may help “scrub” the walls, reducing the rate of deposition of
solids. The presence of acoustic irradiation appeared to enhance the reaction rate, which
may be the result of the energy emitted from cavitations (and thus increased temperature)
and/or enhanced mixing. It was demonstrated that in the presence of acoustics and solids,
more bubble formation occurs, leading to a tighter residence time distribution. Moreover,
in silicon microreactors, the temperature within the device at steady state in the presence
of acoustics was demonstrated to be 1.5ºC higher than the setpoint determined at reactor
surface.
Combining a spiral-layout microreactor performing sequential addition and a biphasic
quench with acoustic irradiation allowed continuous-flow synthesis of the biaryl amine
with NaCl solid byproduct. The reaction was performed at 0.1 M of the aryl chloride
reagent, allowing the synthesis of approximately 0.2 g/hr of the product, which heretofore
has not been possible. Best performance regarding resistance to clogging was observed
at higher flow rates. Application of a constriction model demonstrated that increasing the
flow velocity can reduce the rate of constriction. Based on the results reported herein,
general guidelines can be prescribed to handle salt byproducts during reactions in
microsystems. It is important to limit the particle sizes by removing them from the
reactor before aspect ratios less than 9 are achieved or by imposing an external force such
as acoustic irradiation. The filtration of solvents and reagent mixtures can also eliminate
potential bridging. Moreover, constriction due to wall deposition may be avoided by
171
increasing the flow velocity and by applying ultrasound. Otherwise, estimation of a
constriction rate is useful in determining how often the reactor should be flushed with
pure solvent to remove deposits.
The principles developed here could be extended to other palladium-catalyzed C-N
bond formation reactions. Consequently, laboratory-scale study of production of fine
chemicals prepared with these reactions is possible. Furthermore, microchemical systems
may be applied to screen the optimal ligands and reaction conditions for individual
reactions and complex synthetic pathways, as well as to analyze the reaction kinetics and
mechanisms of important transformations.
Although the ability to flow solids-containing slurries in microsystems expands the
potential use of microreactors for synthetic chemistry, a number of challenges remain.
Pharmaceutical synthesis routes may involve solids participating as reactants or catalysts,
in addition to products or byproducts. Strategies for handling other types of solids will
prove useful in developing the most efficient synthetic pathways and in performing
efficient reaction studies.
172
Chapter 7.
Summary and Future Outlook
7.1
Thesis Contributions
The present thesis was aimed at applying the advancements and developments in the
field of microfluidics to study, accelerate, and enhance industrially interesting but
difficult-to-perform chemical syntheses.
The first half of the thesis detailed the
development of rapid and inexpensive methods of silicon etching, as well as the design of
highly efficient micromixing devices. The second half of the thesis demonstrated the
application of these and other devices to safely and efficiently obtain detailed kinetics
and accelerate reactions that involve extreme or difficult-to-apply conditions or
hazardous intermediates and products.
In Chapter 2, wet etch techniques for silicon device fabrication were developed.
These techniques were designed to replace the existing deep reactive ion etch method,
which is very slow, can be applied to only one wafer at a time, and is rather expensive.
Applying knowledge of the crystalline structure of silicon and of the etch rates of
different crystal planes, a potassium hydroxide etch was developed to enable in a single
etch step to produce features of different depths, as well as through-etched features. This
method can be applied to up to 13 wafers at a time, with high etch consistency across and
among wafers. To allow a greater flexibility of layout design and more physically robust
devices, a wet etch step based on a mixture of nitric and hydrofluoric acids was
developed. This method can be a applied with etch rates of 100 μm/min, two orders of
magnitude faster than the deep reactive ion etch method.
In Chapter 3, a new design of high-flow-rate rapid micromixers was developed.
Based on the concept of laminar interdigitation (splitting reagent flows and interspersing
them in very narrow lamina to take advantage of mixing by diffusion in laminar flow),
the micromixer was designed to effect mixing on the time scale of 1-10 ms, thus enabling
the study of very rapid chemical kinetics by removing liquid mass transfer limitations.
173
The design included both a two-stream and a novel three-stream design, the latter being
capable of ordering the three reagents to produce mixing in a desired sequence. This
design took advantage of compression packaging methods to produce this rapid mixing
with a very low pressure drop of 1×10-2 bar/Qμ, with Q and μ being flow rate in mL/min
and viscosity in centipoises, respectively. Thus, these mixers boast a very high efficiency
of mixing compared to existing designs.
The micromixers were applied in Chapter 4 to a multi-step synthesis of sodium
nitrotetrazolate via a highly energetic tetrazolediazonium intermediate. The kinetics of
both reaction steps were evaluated with accuracy, obtaining orders of reaction, thermal
dependencies, and the dependency of the nitration step on reaction ionic strength and pH.
Microscale flow chemistry allowed this characterization, which would not have been
feasible on the macroscale due to the evolution of gas (which was able to be removed on
the microscale), or in batch due to the high hazards of explosion of the energetic
intermediate and product. Moreover, the diazotization step, which is extremely rapid,
could only be accurately studies using rapid micromixing to ensure accurate reaction
initiation and termination. Additionally, the demonstration of much safer and more
efficient production-level synthesis was accomplished with the microreactor setup,
generating 4.4 g/hour of the nitrotetrazolate compound in a small lab bench footprint.
In Chapter 5, microreactors were applied to attain high pressures to allow volatile
solvents to remain liquid at high temperatures. This capability was used in a study and
acceleration of β-alcohol formation by epoxide aminolysis. Reactions were shown to be
greatly accelerated from their traditional methods, with two pharmaceutically relevant
compounds, metoprolol and a precursor to indacaterol, synthesized with residence times
decreased by a factor of 30-60 from conventional batch methods. The microreactor
system was also used to perform accelerated reaction screening, as well as enabling a full
kinetic study of the epoxide aminolysis multi-step mechanism using only 5 g each of two
reagents and performing 35 experiments in triplicate in an 8-hour time span.
Chapter 6 describes development of a microreactor-based system capable of
performing in flow a solids-generating reaction. A palladium-catalyzed C-N coupling
reaction between an aryl amine and a substituted aryl halide was used as the model
chemistry.
It was learned that clogging in microreactors occurs by two primary
174
mechanisms: deposition on walls and bridging of the channel by particles in the bulk. A
reactor was designed to minimize the effects of bridging by providing a spiral channel
with a maximized turn radius, a quench for the dispersal of solids into another phase prior
to the outlet, and a sequential addition of one reagent to more evenly distribute the solids
throughout the reactor channel.
Ultrasound was found to disrupt the formation of
agglomerates, greatly reducing the potential for bridging. Combined, the ultrasonicated
reactor was able to perform the coupling chemistry in flow with a yield of 65% and
production of 0.2 g/hour of desired product. This can greatly expand the application of
microfluidics for reaction screening, kinetic study, and optimization of solids-containing
reactions.
7.2
Suggestions for Future Work and Outlook
The acidic wet etch process could be augmented by the application of temperaturecontrolled, well-agitated etchant baths, allowing a more precise handling of etch rates.
This may reduce the etch variation across the wafer and enhance the ability to etch
multiple wafers simultaneously. Silicon-to-silicon fusion bonding can be applied to
produce reactors with nearly circular channels, thus simulating tubular reactors to
minimize dispersion while maintaining all advantages of silicon, such as excellent heat
transfer and compact reactor layout.
To enable solids handling of a broad range of reactions, studies should be performed
to analyze the differences in solid behavior among different categories of solids, such as
mineral salts, small organic crystals, polymers and peptides, and catalyst particles.
Additionally, capability to flow solids into microreactors would enable flow study of
reactions using mobile supported catalysts or solid precursors/reagents. To best prevent
wall deposition, a robust method should be developed of depositing on microreactor
surfaces a coating such as a fluoropolymer that can withstand harsh chemistries and high
temperatures while providing an impermeable film. Combined, these techniques may
enable a comprehensive and robust system capable of performing a myriad of chemical
reactions.
The future of microreactors hinges on integration and automation.237 This thesis has
demonstrated the ability to accurately and efficiently study kinetics of multi-step
175
reactions, as well as to handle multi-phase chemistries. Additionally, modular devices
such as micromixers have been demonstrated, both separately and in flow chemistry
systems. Combining these and other microscale devices with the techniques developed in
this thesis and in other works in the field of microfluidics will allow for the creation of
miniature pilot plants capable of simulating full-scale production units. This will lead to
a much more direct process scaling, as well as to greater efficiency and economy during
process optimization and evaluation.
Combining these techniques with intelligent
control, analytics, and optimization algorithms will lead to safe, rapid, economical, and
environmentally friendly systems to develop optimal production-ready processes from
the basic building blocks of flask-derived reaction steps.
The development of
microfluidic systems and applications in this thesis has advanced the microchemical field
from laboratory studies to industrially applicable reactions and scales, permitting safe,
rapid, and efficient process development of a wide range of syntheses.
176
References ..
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
Senturia, S.D., Microsystem Design. 2001, Boston: Kluwer Academic.
Madou, Marc J., Fundamentals of Microfabrication: The Science of
Miniaturization. 2nd ed. 2002, Boca Raton, Florida: CRC Press.
Manz, A.; Graber, N., and Widmer, H. M., "Miniaturized Total ChemicalAnalysis Systems - a Novel Concept For Chemical Sensing," Sensors and
Actuators B-Chemical 1990. 1(1-6): p. 244-248.
Service, Robert F., "Miniaturization Puts Chemical Plants Where You Want
Them," Science 1998. 282(5388): p. 400.
Manz, A.; Harrison, D. J.; Verpoorte, E. M. J.; Fettinger, J. C.; Ludi, H., and
Widmer, H. M., "Miniaturization of Chemical-Analysis Systems - a Look Into
Next Century Technology or Just a Fashionable Craze," Chimia 1991. 45(4): p.
103-105.
Manz, A.; Miyahara, Y.; Miura, J.; Watanabe, Y.; Miyagi, H., and Sato, K.,
"Design of an Open-Tubular Column Liquid Chromatograph Using Silicon Chip
Technology," Sensors and Actuators B-Chemical 1990. 1(1-6): p. 249-255.
Ehrfeld, W.; Hessel, V., and Lowe, H., Microreactors: New Technology for
Modern Chemistry. 2000, Weinheim, Germany: Wiley-VCH.
Jensen, Klavs F., "Microchemical systems: status, challenges, and opportunities,"
AIChE Journal 1999. 45(10): p. 2051-2054.
Ashmead, J. W.; Blaisdell, C. T.; Johnson, M. H.; Nyquist, J. K.; Perrotto, J. A. ,
and Ryley, J. F., Integrated Chemical Processing Apparatus and Processes for
the Preparation Thereof. Patent 5534328, 1996.
Ashmead, J. W.; Blaisdell, C. T.; Johnson, M. H.; Nyquist, J. K.; Perrotto, J. A. ,
and Ryley, J. F., Integrated Chemical Processing Apparatus and Processes for
the Preparation Thereof. Patent 5960763, 1997.
Ryley, J. F.; Perrotto, J. A.; Oren, M., and Riegert, R. J., Waveguide Sensing
Element for Use in a Sample Medium and Method of Rear-Firing
Electromagnetic Radiation. Patent 5724151, 1998.
Hartman, Ryan L. and Jensen, Klavs F., "Microchemical systems for continuousflow synthesis," Lab on a Chip 2009. 9(17): p. 2495-2507.
Incropera, F. and Dewitt, D., Introduction to heat transfer. 1985. Medium: X;
Size: Pages: 704.
de Mas, N.; Günther, A.; Schmidt, M. A., and Jensen, K. F., "Microfabricated
multiphase reactors for the selective direct fluorination of aromatics," Industrial
& Engineering Chemistry Research 2003. 42(4): p. 698-710.
Murphy, E. R.; Martinelli, J. R.; Zaborenko, N.; Buchwald, S. L., and Jensen, K.
F., "Accelerating reactions with microreactors at elevated temperatures and
pressures: Profiling aminocarbonylation reactions," Angewandte ChemieInternational Edition 2007. 46(10): p. 1734-1737.
Quiram, D.J., Characterization and Systems Integration of Microreactors, in
Chemical Engineering. 2002, Massachusetts Institute of Technology: Cambridge,
MA. p. 327.
177
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
Ajmera, S. K.; Losey, M. W.; Jensen, K. F., and Schmidt, M. A.,
"Microfabricated packed-bed reactor for phosgene synthesis," Aiche Journal
2001. 47(7): p. 1639-1647.
Williamson, Kenneth L., Macroscale and microscale organic experiments. 2nd
ed. 1994, Lexington, Mass.: D.C. Heath and Company.
Goodell, John R.; McMullen, Jonathan P.; Zaborenko, Nikolay; Maloney, Jason
R.; Ho, Chuan-Xing; Jensen, Klavs F.; Porco, John A., and Beeler, Aaron B.,
"Development of an Automated Microfluidic Reaction Platform for
Multidimensional
Screening:
Reaction
Discovery
Employing
Bicyclo[3.2.1]octanoid Scaffolds," The Journal of Organic Chemistry 2009.
74(16): p. 6169-6180.
"MIT-Novartis Center for Continuous Manufacturing,"
http://web.mit.edu/newsoffice/2007/novartis-0928.html 28 September 2007.
Ohashi, H.; Ohura, J.; Tsukakoshi, T., and Shimbo, M. Improved Dielectrically
Isolated Device Integration by Silicon-Wafer Direct Bonding Technique. in
Technical Digest: IEEE International Electron Devices Meeting (IEDM '86).
1986. Los Angeles.
Wallis, G. and Pomerantz, D.I., "Field Assisted Glass-Metal Sealing," Journal of
Applied Physics 1969. 40: p. 3946-3949.
de Mas, N.; Günther, A.; Kraus, T.; Schmidt, M. A., and Jensen, K. F., "Scaledout multilayer gas-liquid microreactor with integrated velocimetry sensors,"
Industrial & Engineering Chemistry Research 2005. 44(24): p. 8997-9013.
Nielsen, O.M.; Arana, L.R.; Baertsch, C.D.; Schmidt, M. A., and Jensen, K. F. A
Thermophotovoltaic Micro-Generator for Portable Power Applications. in
Transducers - 12th International Conference on Solid-State Sensors, Actuators
and Microsystems. 2003. Boston MA: Transducers Research Foundation.
Blackwell, Brandon S., Design, fabrication, and characterization of a micro fuel
processor, in Dept. of Chemical Engineering. 2008, Massachusetts Institute of
Technology: Cambridge, MA. p. 174.
Kralj, J. G.; Sahoo, H. R., and Jensen, K. F., "Integrated continuous microfluidic
liquid-liquid extraction," Lab on a Chip 2007. 7(2): p. 256-263.
Plummer, James D.; Deal, Michael D., and Griffin, Peter B., Silicon VLSI
Technology: Fundamentals, Practice, and Modeling. 1 ed. 2000: Prentice Hall.
817.
de Mas, N.; Guenther, A.; Schmidt, M. A., and Jensen, K. F., "Microfabricated
multiphase reactors for the selective direct fluorination of aromatics," Industrial
& Engineering Chemistry Research 2003. 42(4): p. 698-710.
Ratner, D.M.; Murphy, E.R.; Jhunjhunwala, M.; Snyder, D.A.; Jensen, K.F., and
P. H. Seeberger, "Microreactor-based reaction optimization in organic chemistry Glycosylation as a challenge," Chem. Commun. 2005. 5(5): p. 578-580.
Srinivasan, R.; Hsing, I. M.; Berger, P. E.; Jensen, K. F.; Firebaugh, S. L.;
Schmidt, M. A.; Harold, M. P.; Lerou, J. J., and Ryley, J. F., "Micromachined
reactors for catalytic partial oxidation reactions," Aiche Journal 1997. 43(11): p.
3059-3069.
178
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
Murphy, E. R.; Inoue, T.; Sahoo, H. R.; Zaborenko, N., and Jensen, K. F.,
"Solder-based chip-to-tube and chip-to-chip packaging for microfluidic devices,"
Lab on a Chip 2007. 7: p. 1309-1314.
Microfabrication cost evaluation is based on normalized user charges by MIT's
MTL, available at
http://mtlweb.mit.edu/services/fabrication/machine_charges.html.
Kovacs, G. T. A.; Maluf, N. I., and Petersen, K. E., "Bulk micromachining of
silicon," Proceedings of the IEEE 1998. 86(8): p. 1536-1551.
Peters, L., "Plasma Etch Chemistry: The Untold Story," Semiconductiors
International 1992(May): p. 660-670.
Lärmer, F. and Schilp, P., Method of Anisotropically Etching Silicon. Patent 1994.
Losey, M. W.; Jackman, R. J.; Firebaugh, S. L.; Schmidt, M. A., and Jensen, K.
F., "Design and fabrication of microfluidic devices for multiphase mixing and
reaction," Journal of Microelectromechanical Systems 2002. 11(6): p. 709-717.
Chen, Kuo-Shen; Ayón, Arturo A.; Zhang, Xin, and Spearing, S. Mark, "Effect of
Process Parameters on the Surface Morphology and Mechanical Performance of
Silicon Structures After Deep Reactive Ion Etching (DRIE)," Journal of
Microelectromechanical Systems 2002. 11(3): p. 265-275.
Laermer, F. and Urban, A., "Challenges, developments and applications of silicon
deep reactive ion etching," Microelectronic Engineering 2003. 67-68: p. 349-355.
Seidel, H.; Csepregi, L.; Heuberger, A., and Baumgärtel, H., "Anisotropic Etching
of Crystalline Silicon in Alkaline Solutions," Journal of Electrochemical Society
1990. 137(11): p. 3612-3626.
Bassous, E., "Fabrication of novel three-dimensional microstructures by the
anisotropic etching of (100) and (110) silicon," IEEE Transactions: Electron
Devices 1978. ED-25(October): p. 1178–1185.
Bean, K.E., "Anisotropic etching of silicon," IEEE Transactions: Electron
Devices 1978. ED-25(October): p. 1185-1193.
Hoffmann, Martin; Kopka, Peter; Gross, Torsten, and Voges, Edgar, "Optical
fibre switches based on full wafer silicon micromachining," Journal of
Micromechanics and Microengineering 1999(2): p. 151.
Nilsson, D; Jensen, S, and Menon, A, "Fabrication of silicon molds for polymer
optics," Journal of Micromechanics and Microengineering 2003(4): p. S57.
Enoksson, Peter, "New structure for corner compensation in anisotropic KOH
etching," Journal of Micromechanics and Microengineering 1997(3): p. 141.
Wu, Xian-Ping and Ko, Wen H., "Compensating corner undercutting in
anisotropic etching of (100) silicon," Sensors and Actuators 1989. 18(2): p. 207215.
Zhang, Qingxin; Liu, Litian, and Li, Zhijian, "A new approach to convex comer
compensation for anisotropic etching of (100) Si in KOH," Sensors and Actuators
A 1996. 56: p. 251-254.
Peeters, E., Process Development for 3D Silicon Microstructures, with
Application to Mechanical Sensor Design. 1994, Catholic University of Louvain:
Belgium.
Petersen, Kurt E., "Silicon as a mechanical material," Proceedings of the IEEE
1982. 70(5): p. 420-457.
179
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
Schwartz, B. and Robbins, H., "Chemical Etching of Silicon - II: The System HF,
HNO3, H2O, and HC2C3O2," Journal of Electrochemical Society 1960. 107: p.
108-111.
Sparrow, E. M., "Laminar flow in isosceles triangular ducts," Aiche Journal 1962.
8(5): p. 599-604.
Inoue, T.; Schmidt, M. A., and Jensen, K. F., "Microfabricated multiphase
reactors for the direct synthesis of hydrogen peroxide from hydrogen and
oxygen," Industrial & Engineering Chemistry Research 2007. 46(4): p. 11531160.
Levenspiel, O., Chemical reaction engineering. 3 ed. 1999, New York: Wiley.
Vignes, Alain, "Diffusion in Binary Solutions. Variation of Diffusion Coefficient
with Composition," Industrial & Engineering Chemistry Fundamentals 1966.
5(2): p. 189-199.
Okada, Yasumasa and Tokumaru, Yozo, "Precise determination of lattice
parameter and thermal expansion coefficient of silicon between 300 and 1500 K,"
Journal of Applied Physics 1984. 56(2): p. 314-320.
Schwartz, B. and Robbins, H., "Chemical Etching of Silicon - IV: Etching
Technology," Journal of Electrochemical Society 1976. 123(12): p. 1903.
Shemilt, L. W. and Nagarajan, R., "Liquid diffusivities for the system methanol–
toluene," Canadian Journal of Chemistry 1967. 45(10): p. 1143–1148.
Zaborenko, Nikolay; Murphy, Edward R.; Kralj, Jason G., and Jensen, Klavs F.,
"Synthesis and Kinetics of Highly Energetic Intermediates by Micromixers:
Direct Multistep Synthesis of Sodium Nitrotetrazolate," Industrial & Engineering
Chemistry Research 2010. 49(9): p. 4132–4139.
Zaborenko, Nikolay; Murphy, Edward R.; Kralj, Jason G., and Jensen, Klavs F.
Kinetic Study and Safe Production-Scale Synthesis of Sodium Nitrotetrazolate in
Silicon Microreactor Systems. in AIChE Spring National Meeting. 2009. Tampa,
FL.
Danckwerts, P.V., "The definition and measurement of some characteristics of
mixtures," Applied Scientific Research, Section A 1952. 3: p. 279-296.
Graveson, P.; Branjeberg, J., and Jensen, O.S., "Microfluidics - a review,"
Journal of Micromechanics and Microengineering 1993. 3(4): p. 168-182.
Nam-Trung, Nguyen and Zhigang, Wu, "Micromixers - a review," Journal of
Micromechanics and Microengineering 2005(2): p. R1.
Hessel, Volker; Löwe, Holger, and Schönfeld, Friedhelm, "Micromixers--a
review on passive and active mixing principles," Chemical Engineering Science
2005. 60(8-9): p. 2479-2501.
Yen, B. K. H.; Gunther, A.; Schmidt, M. A.; Jensen, K. F., and Bawendi, M. G.,
"A microfabricated gas-liquid segmented flow reactor for high-temperature
synthesis: The case of CdSe quantum dots," Angewandte Chemie-International
Edition 2005. 44(34): p. 5447-5451.
Wootton, R. C. R.; Fortt, R., and de Mello, A. J., "On-chip generation and
reaction of unstable intermediates-monolithic nanoreactors for diazonium
chemistry: Azo dyes," Lab on a Chip 2002. 2(1): p. 5-7.
180
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
Garofalo, F.; Adrover, A.; Cerbelli, S., and Giona, M., "Spectral characterization
of static mixers. The S-shaped micromixer as a case study," AIChE Journal.
56(2): p. 318-335.
Wootton, Robert C. R.; Fortt, Robin, and de Mello, Andrew J., "A
Microfabricated Nanoreactor for Safe, Continuous Generation and Use of Singlet
Oxygen," Organic Process Research & Development 2002. 6(2): p. 187-189.
Jackman, R. J.; Floyd, T. M.; Ghodssi, R.; Schmidt, M. A., and Jensen, K. F.,
"Microfluidic systems with on-line UV detection fabricated in photodefinable
epoxy," Journal of Micromechanics and Microengineering 2001. 11(3): p. 263269.
Hessel, V.; Hardt, S.; Lowe, H., and Schonfeld, F., "Laminar mixing in different
interdigital micromixers: I. Experimental characterization," AIChE Journal 2003.
49(3): p. 566-577.
Hardt, S. and Schonfeld, F., "Laminar mixing in different interdigital
micromixers: II. Numerical simulations," AIChE Journal 2003. 49(3): p. 578-583.
Tilton, James N. , Fluid and Particle Dynamics, in Perry's Chemical Engineers'
Handbook, R.H. Perry and D.W. Green, Editors. 1997, McGraw-Hill: Princeton,
NJ. p. (6-1)-(6-54).
Owen, Transactions of the American Society of Civil Engineers 1954. 119: p.
1157-1175.
Sparrow, E.M., "Laminar Flow in Isosceles Triangular Ducts," AIChE Journal
1962. 8: p. 599-604.
Aubin, Joëlle; Ferrando, Montse, and Jiricny, Vladimir, "Current methods for
characterising mixing and flow in microchannels," Chemical Engineering
Science. 65(6): p. 2065-2093.
Bourne, J.R.; Kut, O.M., and Lenzner, J., "An improved reaction system to
investigate micromixing in high-intensity mixers," Industrial & Engineering
Chemistry Research 1992. 31: p. 949-958.
Fournier, M. C.; Falk, L., and Villermaux, J., "A new parallel competing reaction
system for assessing micromixing efficiency--Experimental approach," Chemical
Engineering Science 1996. 51(22): p. 5053-5064.
Guichardon, Pierrette and Falk, Laurent, "Characterisation of micromixing
efficiency by the iodide-iodate reaction system. Part I: experimental procedure,"
Chemical Engineering Science 2000. 55(19): p. 4233-4243.
Villermaux, J.; Falk, L., and Fournier, M. C., "Use of parallel competing reactions
to characterize micromixing efficiency," AIChE Symposium Series 1992. 88: p.
286-290.
Villermaux, J.; Falk, L., and Fournier, M. C., "Potential use of a new parallel
reaction system to characterize micromixing in stirred reactors," AIChE
Symposium Series 1994. 90: p. 299-304.
Aoki, Nobuaki and Mae, Kazuhiro, "Effects of channel geometry on mixing
performance of micromixers using collision of fluid segments," Chemical
Engineering Journal 2006. 118(3): p. 189-197.
Keoschkerjan, R.; Richter, M.; Boskovic, D.; Schnürer, F., and Löbbecke, S.,
"Novel multifunctional microreaction unit for chemical engineering," Chemical
Engineering Journal 2004. 101(1-3): p. 469-475.
181
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
Kockmann, Norbert; Kiefer, Thomas; Engler, Michael, and Woias, Peter,
"Convective mixing and chemical reactions in microchannels with high flow
rates," Sensors and Actuators B: Chemical 2006. 117(2): p. 495-508.
Nagasawa, H.; Aoki, N., and Mae, K., "Design of a New Micromixer for Instant
Mixing Based on the Collision of Micro Segments," Chemical Engineering &
Technology 2005. 28(3): p. 324-330.
Panić, S.; Loebbecke, S.; Tuercke, T.; Antes, J., and Boskovic, D., "Experimental
approaches to a better understanding of mixing performance of microfluidic
devices," Chemical Engineering Journal 2004. 101(1-3): p. 409-419.
Jensen, K. F., "Microreaction engineering - is small better?," Chemical
Engineering Science 2001. 56(2): p. 293-303.
Ratner, Daniel Martin, Solution-phase and automated solid-phase synthesis of
high-mannose oligosaccharides : application to carbohydrate microarrays and
biological studies, in Chemistry. 2004, Massachusetts Institute of Technology:
Cambridge, MA. p. 355.
Minegishi, Yutaka; Tsukamasa, Yasuyuki; Katayama, Kazunori, and Imai,
Chiharu, Antimicrobial agents containing diazonium compounds. Patent JP.
08,310,957 [96,310,957], 1996.
Peppercorn, Mark A., "Sulfasalazine. Pharmacology, clinical use, toxicity, and
related new drug development," Annals of Internal Medicine 1984. 101(3): p.
377-86.
Fiedler H, Hahn von Dorsche H, "5-Diazonium-1 H-tetrazole--a new reagent for
the histochemical demonstration of histidine," Acta Histochemica 1970. 35(2): p.
414-6.
Murphy, Amanda R.; St. John, Peter, and Kaplan, David L., "Modification of silk
fibroin using diazonium coupling chemistry and the effects on hMSC proliferation
and differentiation " Biomaterials 2008. 29(19): p. 2829-2838.
Fronabarger, John W.; Williams, Michael D., and Sanborn, William B., Jr.
Characterization and Output Testing of the Novel Primary Explosive,
Bis(furoxano)nitrophenol, Potassium Salt. in 41st AIAA/ASME/SAE/ASEE Joint
Propulsion Conference & Exhibit. 2005. Tucson, Arizona.
Klapötke, Thomas M. ; Sabaté, Carles Miró , and Welch, Jan M., "Alkali metal 5nitrotetrazolate salts: prospective replacements for service lead(II) azide in
explosive initiators," Dalton Transactions 2008: p. 6372 - 6380.
Fortt, R.; Wootton, R. C. R., and de Mello, A. J., "Continuous-flow generation of
anhydrous diazonium species: Monolithic microfluidic reactors for the chemistry
of unstable intermediates," Organic Process Research & Development 2003. 7(5):
p. 762-768.
Günther, P. M.; Möller, F.; Henkel, T.; Köhler, J. M., and Groß, G. A.,
"Formation of monomeric and novolak azo dyes in nanofluid segments by use of
a double injector chip reactor," Chemical Engineering & Technology 2005. 28(4):
p. 520-527.
Morosin, B.; Dunn, R. G.; Assink, R.; Massis, T. M.; Fronabarger, J., and
Duesler, E. N., "The secondary explosive tetraammine-cis-bis(5-nitro-2Htetrazolato-N2)cobalt(III) perchlorate at 293 and 213 K," Acta Crystallographica
Section C-Crystal Structure Communications 1997. 53: p. 1609-1611.
182
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
Bubnov, P. F., Primary Explosives And Initiation Devices. 1940, Ministry of
Defence Industry Press: Moscow. p. 311-312.
Thiele, Johannes and Marais, J. T., "Tetrazolderivate aus Diazotetrazotsäure,"
Justus Liebig's Annalen der Chemie 1893. 273(2-3): p. 144-160.
Fronabarger, John; Schuman, Alex; Chapman, Robert D.; Fleming, Wayne, and
Sanborn, William B. Chemistry and Development of BNCP, A Novel DDT
Explosive. in 31st AIAA-Joint Propulsion Conference. 1995. San Diego, CA:
American Institute of Aeronautics and Astronautics Inc.
Galli, C., "Substituent Effects on the Sandmeyer Reaction - Quantitative Evidence
for Rate-Determining Electron-Transfer," Journal of the Chemical Society-Perkin
Transactions 2 1984(5): p. 897-902.
Gilligan, W. H. and Kamlet, M. J., Method of Preparing The Acid Copper Salt of
5-Nitrotetrazole. Patent 4,093,623, 1978.
Koldobskii, G.I.; Soldatenko, D.S.; Gerasimova, E.S.; Hohrjakova, M.B.;
Sherbin, M.B.; Lebedev, V.P., and Ostrovskii, V.A., "Tetrazoles: XXXVI
Synthesis, structure and properties of 5-nitrotetrazole," Russian Journal of
Organic Chemistry 1997. 33(12): p. 1771-1783.
Sahoo, Hemantkumar, Design and Operation of Microchemical Systems for
Multistep Chemical Syntheses, in Chemical Engineering. 2008, MIT: Cambridge,
MA. p. 184.
Hegarty, A.F., Kinetics and mechanisms of reactions involving diazonium and
diazo groups, in The chemistry of diazonium and diazo groups, S. Patai, Editor.
1978, Wiley: Chichester.
Hammett, L.P., Physical Organic Chemistry. 1940, New York: McGraw-Hill.
294.
Bunton, C.A.; D.R., Llewellyn, and Stedman, G., "Oxygen exchange between
nitrous acid and water," Journal of the Chemical Society (Resumed) 1959: p. 568572.
de Fabrizio, E.C.R; Kalatzis, E., and Ridd, J.H., "Nitrosation, Diazotisation, and
Deamination. Part XIII. Substituent Effects of the Nitrosation of Anilinium Ions,"
Journal of the Chemical Society B: Physical Organic 1966: p. 533-539.
Debye, P. and Hückel, E., "The theory of electrolytes. I. Lowering of freezing
point and related phenomena," Physikalische Zeitschrift 1923. 24: p. 185-206.
Meissner, H. P. and Tester, J. W., "Activity coefficients of strong electrolytes in
aqueous solutions," Industrial & Engineering Chemistry Process Design and
Development 1972. 11(1): p. 128-133.
Chen, Chau-Chyun; Britt, H. I.; Boston, J. F., and Evans, L. B., "Local
composition model for excess Gibbs energy of electrolyte systems. Part I: Single
solvent, single completely dissociated electrolyte systems," AIChE Journal 1982.
28(4): p. 588-596.
Hamann, Carl H.; Hamnett, Andrew, and Vielstich, Wolf, Electrochemistry. 2 ed.
2007, Weinheim Wiley-VCH. 2.5.2.
Bedore, Matthew W.; Zaborenko, Nikolay; Jensen, Klavs F., and Jamison,
Timothy F., "Aminolysis of Epoxides in a Microreactor System: A Continuous
Flow Approach to β-Amino Alcohols," Organic Process Research &
Development 2010. 14(2): p. 432-440.
183
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
Bedore, Matthew W.; Zaborenko, Nikolay; Jensen, Klavs F., and Jamison,
Timothy F., Continuous Flow Synthesis of Amino Alcohols Using Microreactors.
Patent Application Number US 61/258,140, 2009.
Bergmeier, Stephen C., "The Synthesis of Vicinal Amino Alcohols," Tetrahedron
2000. 56(17): p. 2561-2576.
Cepanec, Ivica; Litvić, Mladen; Mikuldaš, Hrvoje; Bartolinčić, Anamarija, and
Vinković, Vladimir, "Calcium trifluoromethanesulfonate-catalysed aminolysis of
epoxides," Tetrahedron 2003. 59(14): p. 2435-2439.
Chini, Marco; Crotti, Paolo; Favero, Lucilla; Macchia, Franco, and Pineschi,
Mauro, "Lanthanide(III) trifluoromethanesulfonates as extraordinarily effective
new catalysts for the aminolysis of 1,2-epoxides," Tetrahedron Letters 1994.
35(3): p. 433-436.
Procopio, Antonio; Gaspari, Marco; Nardi, Monica; Oliverio, Manuela, and
Rosati, Ornelio, "Highly efficient and versatile chemoselective addition of amines
to epoxides in water catalyzed by erbium(III) triflate," Tetrahedron Letters 2008.
49(14): p. 2289-2293.
Yadav, J. S.; Reddy, A. Ramesh; Narsaiah, A. Venkat, and Reddy, B. V. S., "An
efficient protocol for regioselective ring opening of epoxides using samarium
triflate: Synthesis of propranolol, atenolol and RO363," Journal of Molecular
Catalysis A: Chemical 2007. 261(2): p. 207-212.
Cossy, Janine; Bellosta, Véronique; Hamoir, Claire, and Desmurs, Jean-Roger,
"Regioselective ring opening of epoxides by nucleophiles mediated by lithium
bistrifluoromethanesulfonimide," Tetrahedron Letters 2002. 43(39): p. 70837086.
Desai, Hinal; D'Souza, Brendan R.; Foether, Devin; Johnson, Benjamin F., and
Lindsay, Harriet A., "Regioselectivity in a Highly Efficient, Microwave-Assisted
Epoxide Aminolysis," Synthesis 2007. 2007(06): p. 902-910.
Maheswara, Muchchintala; Rao, Kummari Subba Venkata Krishna, and Do, Jung
Yun, "Regioselective ring-opening of epoxides with amines using Zn(ClO4)2Al2O3 as a heterogeneous and recyclable catalyst," Tetrahedron Letters 2008.
49(11): p. 1795-1800.
Onaka, Makoto; Kawai, Motomitsu, and Izumi, Yusuke, "Zeolite-Catalyzed RingOpening Of Epoxides With Amines," Chemistry Letters 1985. 14(6): p. 779-782.
Vijender, Medamoni; Kishore, P.; Narender, P., and Satyanarayana, B.,
"Amberlist-15 as heterogeneous reusable catalyst for regioselective ring opening
of epoxides with amines under mild conditions," Journal of Molecular Catalysis
A: Chemical 2007. 266(1-2): p. 290-293.
Ramesh Kumar, S. and Leelavathi, P., "Phosphomolybdic acid-Al2O3: A mild,
efficient, heterogeneous and reusable catalyst for regioselective opening of
oxiranes with amines to [beta]-amino alcohols," Journal of Molecular Catalysis
A: Chemical 2007. 266(1-2): p. 65-68.
Azizi, Najmodin; Torkiyan, Lalleh, and Saidi, Mohammad R., "Highly Efficient
One-Pot Three-Component Mannich Reaction in Water Catalyzed by Heteropoly
Acids," Organic Letters 2006. 8(10): p. 2079-2082.
184
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
Bonollo, Simona; Fringuelli, Francesco; Pizzo, Ferdinando, and Vaccaro, Luigi,
"A green route to β-amino alcohols via the uncatalyzed aminolysis of 1,2epoxides by alkyl- and arylamines," Green Chemistry 2006. 8(11): p. 960-964.
Kravchenko, V.V.; Kostenko, L. I.; Popov, A.F.; Kotenko, A.A., and Koblik, I.
V., "Solvent Effect on the Reaction Rate of Aminolysis of Substituted α-Oxides,"
Ukrainian Chemistry Journal 1990. 56(2): p. 168-172.
Vinnik, R.M. and Roznyatovsky, V.A., "Kinetic Method by Using Calorimetry to
Mechanism of Epoxy-Amine Cure Reaction; Part I. Mangeldorf's Approach,"
Journal of Thermal Analysis and Calorimetry 2003. 73: p. 807-817.
Vinnik, R.M. and Roznyatovsky, V.A., "Kinetic Method by Using Calorimetry to
Mechanism of Epoxy-Amine Cure Reaction; Part II. On catalytic action of the
amine excess," Journal of Thermal Analysis and Calorimetry 2003. 73: p. 819826.
Vinnik, R.M. and Roznyatovsky, V.A., "Kinetic Method by Using Calorimetry to
Mechanism of Epoxy-Amine Cure Reaction; Part V. Phenyl glycidyl ether aniline.," Journal of Thermal Analysis and Calorimetry 2004. 75: p. 753-764.
Sirovski, Felix; Mulyashov, Sergei, and Shvets, Valery, "Large-scale fatty amine
ethoxylation reactor: A dynamic model," Chemical Engineering Journal 2006.
117(3): p. 197-203.
Sundaram, P.K. and Sharma, M.M., "Kinetics of Reactions of Amines with
Alkene Oxides," Bulletin of the Chemical Society of Japan 1969. 42(11): p. 31413147.
Rogers, Donald Z. and Bruice, Thomas C., "Comparative chemistry of the bayand non-bay-region tetrahydro epoxides of phenanthrene," Journal of the
American Chemical Society 1979. 101(16): p. 4713-4719.
Trejbal, Jiří and Petrisko, Miroslav, "Kinetics of ethylenediamine and piperazine
ethoxylation," Reaction Kinetics and Catalysis Letters 2004. 82(2): p. 339-346.
Lidström, Pelle; Tierney, Jason; Wathey, Bernard, and Westman, Jacob,
"Microwave assisted organic synthesis--a review," Tetrahedron 2001. 57(45): p.
9225-9283.
Robin, Aélig; Brown, Fraser; Bahamontes-Rosa, Noemí; Wu, Binghua; Beitz,
Eric; Kun, Jürgen F. J., and Flitsch, Sabine L., "Microwave-Assisted Ring
Opening of Epoxides: A General Route to the Synthesis of 1-Aminopropan-2-ols
with Anti Malaria Parasite Activities," Journal of Medicinal Chemistry 2007.
50(17): p. 4243-4249.
Kappe, C. Oliver, "Controlled Microwave Heating in Modern Organic Synthesis,"
Angewandte Chemie International Edition 2004. 43(46): p. 6250-6284.
Bowman, Matthew D.; Holcomb, Jennifer L.; Kormos, Chad M.; Leadbeater,
Nicholas E., and Williams, Victoria A., "Approaches for Scale-Up of MicrowavePromoted Reactions," Organic Process Research & Development 2007. 12(1): p.
41-57.
Bowman, Matthew D.; Schmink, Jason R.; McGowan, Cynthia M.; Kormos,
Chad M., and Leadbeater, Nicholas E., "Scale-Up of Microwave-Promoted
Reactions to the Multigram Level Using a Sealed-Vessel Microwave Apparatus,"
Organic Process Research & Development 2008. 12(6): p. 1078-1088.
185
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
Glasnov, Toma N. and Kappe, C. Oliver, "Microwave-Assisted Synthesis under
Continuous-Flow Conditions," Macromolecular Rapid Communications 2007.
28(4): p. 395-410.
Moseley, Jonathan D. and Woodman, Emily K., "Scaling-Out Pharmaceutical
Reactions in an Automated Stop-Flow Microwave Reactor," Organic Process
Research & Development 2008. 12(5): p. 967-981.
Benito-López, Fernando; Egberink, Richard J. M.; Reinhoudt, David N., and
Verboom, Willem, "High pressure in organic chemistry on the way to
miniaturization," Tetrahedron 2008. 64(43): p. 10023-10040.
Razzaq, Tahseen; Glasnov, Toma N., and Kappe, C. Oliver, "Continuous-Flow
Microreactor Chemistry under High-Temperature/Pressure Conditions,"
European Journal of Organic Chemistry 2009. 2009(9): p. 1321-1325.
Jas, Gerhard and Kirschning, Andreas, "Continuous flow techniques in organic
synthesis," Chemistry--A European Journal 2003. 9(23): p. 5708-5723.
Mason, Brian P.; Price, Kristin E.; Steinbacher, Jeremy L.; Bogdan, Andrew R.,
and McQuade, D. Tyler, "Greener Approaches to Organic Synthesis Using
Microreactor Technology," Chemical Reviews 2007. 107(6): p. 2300-2318.
Wiles, Charlotte and Watts, Paul, "Improving chemical synthesis using flow
reactors," Expert Opinion on Drug Discovery 2007. 2(11): p. 1487-1503.
Wiles, Charlotte and Watts, Paul, "Continuous Flow Reactors, a Tool for the
Modern Synthetic Chemist," European Journal of Organic Chemistry 2008.
2008(10): p. 1655-1671.
Roberge, Dominique M.; Zimmermann, Bertin; Rainone, Fabio; Gottsponer,
Michael; Eyholzer, Markus, and Kockmann, Norbert, "Microreactor Technology
and Continuous Processes in the Fine Chemical and Pharmaceutical Industry: Is
the Revolution Underway?," Organic Process Research & Development 2008.
12(5): p. 905-910.
Tachibana, Shinya; Yoshiyama, Ryuji; Oishi, Tsuyoshi; Sakurai, Mikiya; Ishida,
Kazuo; Tsujiuchi, Tatsuya; Mita, Hidehisa; Araki, Ryosuke, and Saito, Kenji,
Process And Equipment For The Production Of Mono(Lower
Alkyl)Monoalkanolamines. Patent WO/2008/068927, 2007.
Cuenoud, Bernard; Bruce, Ian; Fairhurst, Robin Alec, and Beattie, David, β2Adrenoceptor Agonists. Patent WO/2000/075114, 12/14/2000, 2000.
Sturton, Richard G.; Trifilieff, Alexandre; Nicholson, Andrew G., and Barnes,
Peter J., "Pharmacological Characterization of Indacaterol, a Novel Once Daily
Inhaled β2 Adrenoceptor Agonist, on Small Airways in Human and Rat PrecisionCut Lung Slices," Journal of Pharmacology and Experimental Therapeutics 2008.
324(1): p. 270-275.
Lohse, Olivier and Vogel, Caspar, Process For Preparing 5-‘(R)-2-(5,6-DiethylIndian-2-Ylamino)-1-Hydroxy-Ethyl!-8-Hydroxy-(1h)-Quinolin-2-One
Salt,
Useful As An Adrenoceptor Agonist. Patent WO/2004/076422, 9/10/2004, 2004.
Waagstein, F.; Hjalmarson, A.; Swedberg, K.; Bristow, M. R.; Gilbert, E. M.;
Camerini, F.; Fowler, M. B.; Silver, M. A.; Johnson, M. R., and Goss, F. G.,
"Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy," The
Lancet 1993. 342(8885): p. 1441-1446.
186
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
Mehra, Janakraj Karamchand; Choubey, Ajit; Srivastava, Bimal Kumar; Porwal,
Rajendra Kumar, and Gautam, Prashant, Metoprolol Manufacturing Process.
Patent US 2005/0107635 A1, Mar 23, 2004, 2005.
Palmer, Sven and Sidenqvist, Michael, New Manufacturing Process of
Metoprolol. Patent WO/1998/022426, 5/28/1998, 1998.
Rao, A. V. Rama; Gurjar, M. K., and Joshi, Shreerang V., A New Route to (±)
Metoprolol. Patent 139:230463, 1997.
Arnalot Aguilar, Carmen and Bosch I Lladó, Jordi, Process for the Preparation
of Metoprolol and its Salts. Patent WO/2007/141593, 12/13/2007, 2007.
Marre, Samuel; Park, Jongnam; Rempel, Jane; Guan, Juan; Bawendi, Moungi G.,
and Jensen, Klavs F., "Supercritical Continuous-Microflow Synthesis of Narrow
Size Distribution Quantum Dots," Advanced Materials 2008. 20(24): p. 48304834.
Jensen, K. F., "Silicon-based microchemical systems: Characteristics and
applications," Materials Research Society Bulletin 2006. 31(2): p. 101-107.
Yen, Brian; Guenther, Axel; Jensen, K. F; Bawendi, Moungi G., and Schmidt, M.
A., Flow method and reactor for manufacturing noncrystals. Patent 7316967, Jan
8, 2008, 2008.
Morini, Gian Luca, "Single-phase convective heat transfer in microchannels: a
review of experimental results," International Journal of Thermal Sciences 2004.
43(7): p. 631-651.
Park, Hee Sung and Punch, Jeff, "Friction factor and heat transfer in multiple
microchannels with uniform flow distribution," International Journal of Heat and
Mass Transfer 2008. 51(17-18): p. 4535-4543.
Deen, WM, Analysis of Transport Phenomena. 1998: Oxford.
Wu, H. Y. and Cheng, Ping, "An experimental study of convective heat transfer in
silicon microchannels with different surface conditions," International Journal of
Heat and Mass Transfer 2003. 46(14): p. 2547-2556.
Beeley, Lee James and Dean, David Kenneth, Novel Heterocyclic Ethanolamine
Derivatives
With
Beta-Adrenoreceptor
Agonistic
Activity.
Patent
PCT/EP1995/000794, 09/21/1995, 1995.
Lohse, Olivier; Vogel, Caspar, and Penn, Gerhard, Enantioselective Preparation
of Quinoline Derivative. Patent WO/2005/123684, 12/29/2005, 2005.
Mammen, Mathai and Mischki, Trevor, Compounds Having β2 Adrenergic
Receptor Agonist And Muscarinic Receptor Antagonist Activity. Patent
WO/2006/023457, 03/02/2006, 2006.
Prashad, Mahavir; Hu, Bin; Har, Denis; Repič, Oljan; Blacklock, Thomas J., and
Lohse, Olivier, "An Efficient and Economical Synthesis of 5,6-Diethyl-2,3dihydro-1H-inden-2-amine Hydrochloride," Organic Process Research &
Development 2005. 10(1): p. 135-141.
Anderson, Shelby R.; Ayers, Joshua T.; DeVries, Keith M.; Fumitaka, Ito;
Mendenhall, Debra, and Vanderplas, Brian C., "The preparation of β-substituted
amines from mixtures of epoxide opening products via a common aziridinium ion
intermediate," Tetrahedron: Asymmetry 1999. 10(14): p. 2655-2663.
Young, Hugh D., Statistical Treatment of Experimental Data. 1962, New York:
McGraw-Hill.
187
169.
170.
171.
172.
173.
174.
175.
176.
177.
178.
179.
180.
181.
182.
183.
184.
185.
Parker, R. E. and Isaacs, N. S., "Mechanisms Of Epoxide Reactions," Chemical
Reviews 1959. 59(4): p. 737-799.
Myint, San; Daud, Wan; Mohamad, Abu, and Kadhum, Abdul, "Temperaturedependent diffusion coefficient of soluble substances during ethanol extraction of
clove," Journal of the American Oil Chemists' Society 1996. 73(5): p. 603-610.
Sudarsan, Arjun P. and Ugaz, Victor M., "Multivortex micromixing,"
Proceedings of the National Academy of Sciences 2006. 103(19): p. 7228-7233.
Johnson, Mark and Kamm, Roger D., "Numerical studies of steady flow
dispersion at low Dean number in a gently curving tube," Journal of Fluid
Mechanics Digital Archive 1986. 172(-1): p. 329-345.
Carey, J. S.; Laffan, D.; Thomson, C., and Williams, M. T., "Analysis of the
reactions used for the preparation of drug candidate molecules," Organic &
Biomolecular Chemistry 2006. 4(12): p. 2337-2347.
Dugger, R. W.; Ragan, J. A., and Ripin, D. H. B., "Survey of GMP bulk reactions
run in a research facility between 1985 and 2002," Org. Proc. Res. Dev. 2005.
9(3): p. 253-258.
Pennemann, H.; Watts, P.; Haswell, S. J.; Hessel, V., and Lowe, H.,
"Benchmarking of microreactor applications," Organic Process Research &
Development 2004. 8(3): p. 422-439.
Fletcher, P. D. I.; Haswell, S. J.; Pombo-Villar, E.; Warrington, B. H.; Watts, P.;
Wong, S. Y. F., and Zhang, X. L., "Micro reactors: principles and applications in
organic synthesis," Tetrahedron 2002. 58(24): p. 4735-4757.
Hessel, V. and Lowe, H., "Organic synthesis with microstructured reactors,"
Chemical Engineering & Technology 2005. 28(3): p. 267-284.
Watts, P. and Wiles, C., "Synthesis of analytically pure compounds in flow
reactors," Chemical Engineering & Technology 2007. 30(3): p. 329-333.
de Mello, Andrew J., "Control and detection of chemical reactions in microfluidic
systems," Nature 2006. 422: p. 394-402.
Jensen, K. F., Microchemical systems for discovery and development, in New
Avenues to Effcient Chemical Synthesis: Emerging Technologies, P.H. Seeberger
and T. Blume, Editors. 2007, Springer-Verlag Berlin: Heidelberg. p. 57-76.
Hartman, R. L. and Jensen, K. F., "Microchemical systems for continuous-flow
synthesis," Lab Chip 2009. 9: p. 2495–2507.
Jähnisch, K.; Hessel, V.; Lowe, H., and Baerns, M., "Chemistry in
microstructured reactors," Angewandte Chemie-International Edition 2004. 43(4):
p. 406-446.
Watts, P. and Wiles, C., "Recent advances in synthetic micro reaction
technology," Chem. Commun. 2007(5): p. 443-467.
Baxendale, I. R.; Deeley, J.; Griffiths-Jones, C. M.; Ley, S. V.; Saaby, S., and
Tranmer, G. K., "A flow process for the multi-step synthesis of the alkaloid
natural product oxomaritidine: a new paradigm for molecular assembly,"
Chemical Communications 2006(24): p. 2566-2568.
Haswell, S. J.; O'Sullivan, B., and Styring, P., "Kumada-Corriu reactions in a
pressure-driven microflow reactor," Lab on a Chip 2001. 1(2): p. 164-166.
188
186.
187.
188.
189.
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
200.
201.
Losey, M. W.; Schmidt, M. A., and Jensen, K. F., "Microfabricated multiphase
packed-bed reactors: Characterization of mass transfer and reactions," Industrial
& Engineering Chemistry Research 2001. 40(12): p. 2555-2562.
Trachsel, F.; Tidona, B.; Desportes, S., and Rudolf von Rohr, Ph, "Solid catalyzed
hydrogenation in a Si/glass microreactor using supercritical CO2 as the reaction
solvent," J. Supercrit. Fluids 2009. 48(2): p. 146-153.
Wiles, C.; Watts, P., and Haswell, S. J., "The use of solid-supported reagents for
the multi-step synthesis of analytically pure alpha,beta-unsaturated compounds in
miniaturized flow reactors," Lab Chip 2007. 7(3): p. 322-330.
Conant, Travis; Karim, Ayman; Rogers, Stephen; Samms, Stephen; Randolph,
Gerard, and Datye, Abhaya, "Wall coating behavior of catalyst slurries in nonporous ceramic microstructures," Chem. Eng. Sci. 2006. 61(17): p. 5678-5685.
Kobayashi, J.; Mori, Y.; Okamoto, K.; Akiyama, R.; Ueno, M.; Kitamori, T., and
Kobayashi, S., "A microfluidic device for conducting gas-liquid-solid
hydrogenation reactions," Science 2004. 304(5675): p. 1305-1308.
Khan, S. A.; Gunther, A.; Schmidt, M. A., and Jensen, K. F., "Microfluidic
synthesis of colloidal silica," Langmuir 2004. 20(20): p. 8604-8611.
Khan, S. A. and Jensen, K. F., "Microfluidic synthesis of titania shells on
colloidal silica," Advanced Materials 2007. 19: p. 2556-+.
Takagi, M.; Maki, T.; Miyahara, M., and Mae, K., "Production of titania
nanoparticles by using a new microreactor assembled with same axle dual pipe,"
Chem. Eng. J. 2004. 101(1-3): p. 269-276.
Poe, S. L.; Cummings, M. A.; Haaf, M. R., and McQuade, D. T., "Solving the
clogging problem: Precipitate-forming reactions in flow," Angew. Chem. Int. Edit.
2006. 45(10): p. 1544-1548.
Song, H.; Chen, D. L., and Ismagilov, R. F., "Reactions in droplets in
microflulidic channels," Angew. Chem. Int. Edit. 2006. 45(44): p. 7336-7356.
Shestopalov, I.; Tice, J. D., and Ismagilov, R. F., "Multi-step synthesis of
nanoparticles performed on millisecond time scale in a microfluidic droplet-based
system," Lab Chip 2004. 4(4): p. 316-321.
Li, W.; Pharn, H. H.; Nie, Z.; MacDonald, B.; Guenther, A., and Kumacheva, E.,
"Multi-step microfluidic polymerization reactions conducted in droplets: The
internal trigger approach," Journal of the American Chemical Society 2008.
130(30): p. 9935-9941.
Nagasawa, H. and Mae, K., "Development of a new microreactor based on
annular microsegments for fine particle production," Industrial & Engineering
Chemistry Research 2006. 45(7): p. 2179-2186.
Kockmann, N.; Kastner, J., and Woias, P., "Reactive particle precipitation in
liquid microchannel flow," Chem. Eng. J. 2008. 135: p. S110-S116.
Horie, T.; Sumino, M.; Tanaka, T.; Matsushita, Y.; Ichimura, T., and Yoshida, J.,
"Photodimerization of Maleic Anhydride in a Microreactor Without Clogging,"
Org. Proc. Res. Dev. 2010: p. 10.1021/op900306z.
Di Carlo, D.; Irimia, D.; Tompkins, R. G., and Toner, M., "Continuous inertial
focusing, ordering, and separation of particles in microchannels," P. Natl. Acad.
Sci. USA 2007. 104(48): p. 18892-18897.
189
202.
203.
204.
205.
206.
207.
208.
209.
210.
211.
212.
213.
214.
215.
216.
Yamada, M. and Seki, M., "Hydrodynamic filtration for on-chip particle
concentration and classification utilizing microfluidics," Lab Chip 2005. 5(11): p.
1233-1239.
Kuntaegowdanahalli, S. S.; Bhagat, A. A. S.; Kumar, G., and Papautsky, I.,
"Inertial microfluidics for continuous particle separation in spiral microchannels,"
Lab Chip 2009. 9(20): p. 2973-2980.
Huang, L. R.; Cox, E. C.; Austin, R. H., and Sturm, J. C., "Continuous particle
separation through deterministic lateral displacement," Science 2004. 304(5673):
p. 987-990.
Kralj, J. G.; Lis, M. T. W.; Schmidt, M. A., and Jensen, K. F., "Continuous
dielectrophoretic size-based particle sorting," Analytical Chemistry 2006. 78(14):
p. 5019-5025.
Pamme, N., "Continuous flow separations in microfluidic devices," Lab Chip
2007. 7(12): p. 1644-1659.
Laurell, T.; Petersson, F., and Nilsson, A., "Chip integrated strategies for acoustic
separation and manipulation of cells and particles," Chem. Soc. Rev. 2007. 36(3):
p. 492-506.
Nilsson, A.; Petersson, F.; Jonsson, H., and Laurell, T., "Acoustic control of
suspended particles in micro fluidic chips," Lab on a Chip 2004. 4(2): p. 131-135.
Petersson, F.; Aberg, L.; Sward-Nilsson, A. M., and Laurell, T., "Free flow
acoustophoresis: Microfluidic-based mode of particle and cell separation,"
Analytical Chemistry 2007. 79(14): p. 5117-5123.
Petersson, F.; Nilsson, A.; Holm, C.; Jonsson, H., and Laurell, T., "Continuous
separation of lipid particles from erythrocytes by means of laminar flow and
acoustic standing wave forces," Lab on a Chip 2005. 5(1): p. 20-22.
Hartman, R. L.; Naber, J. R.; Zaborenko, N.; Buchwald, S. L., and Jensen, K. F.
Handling solids in microreactors for continuous-flow synthetic chemistry. in
AIChE Annual Conference. 2009. Nashville, TN.
Hartman, R. L.; Naber, J. R.; Zaborenko, N.; McMullen, J. P., and Jensen, K. F.,
Systems and methods for handling solids in microfluidic systems, Massachusetts
Institute of Technology. Patent Application Number US 61/258,767, 2009.
Surry, D. S. and Buchwald, S. L., "Biaryl phosphane ligands in palladiumcatalyzed amination," Angew. Chem. Int. Ed. 2008. 47(34): p. 6338-6361.
Wolfe, J. P.; Wagaw, S.; Marcoux, J. F., and Buchwald, S. L., "Rational
development of practical catalysts for aromatic carbon-nitrogen bond formation,"
Acc. Chem. Res. 1998. 31(12): p. 805-818.
Hartwig, J. F., "Transition metal catalyzed synthesis of arylamines and aryl ethers
from aryl halides and triflates: Scope and mechanism," Angew. Chem. Int. Ed.
1998. 37(15): p. 2047-2067.
Huang, Xiaohua; Anderson, Kevin W.; Zim, Danilo; Jiang, Lei; Klapars, Artis,
and Buchwald, Stephen L., "Expanding Pd-Catalyzed C-N Bond-Forming
Processes: The First Amidation of Aryl Sulfonates, Aqueous Amination, and
Complementarity with Cu-Catalyzed Reactions," Journal of the American
Chemical Society 2003. 125(22): p. 6653-6655.
190
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
229.
230.
231.
232.
233.
Altman, Ryan A.; Fors, Brett P., and Buchwald, Stephen L., "Pd-catalyzed
amination reactions of aryl halides using bulky biarylmonophosphine ligands,"
Nat. Protocols 2007. 2(11): p. 2881-2887.
Ramachandran, V. and Fogler, H. S., "Plugging by hydrodynamic bridging during
flow of stable colloidal particles within cylindrical pores," J. Fluid Mech. 1999.
385: p. 129-156.
Vitthal, S. and Sharma, M. M., "A Stokesian Dynamics Model For Particle
Deposition And Bridging In Granular Media," J. Colloid Interf. Sci. 1992. 153(2):
p. 314-336.
Muecke, T. W., "Formation Fines And Factors Controlling Their Movement In
Porous-Media," J. Petrol. Tech. 1979. 31(2): p. 144-150.
Wyss, Hans M.; Blair, Daniel L.; Morris, Jeffrey F.; Stone, Howard A., and
Weitz, David A., "Mechanism for clogging of microchannels," Phys. Rev. E Stat.
Nonlin. Soft. Matter Phys. 2006. 74(6): p. 061402.
Ramachandran, V. and Fogler, H. S., "Multilayer deposition of stable colloidal
particles during flow within cylindrical pores," Langmuir 1998. 14(16): p. 44354444.
Song, L. F. and Elimelech, M., "Transient Deposition of Colloidal Particles in
Heterogeneous Porous-Media," J. Colloid Interf. Sci. 1994. 167(2): p. 301-313.
Marshall, J. K. and Kitchener, J. A., "The deposition of colloidal particles on
smooth solids," J. Colloid Interf. Sci. 1966. 22(4): p. 342-351.
Shekhar, S.; Ryberg, P.; Hartwig, J. F.; Mathew, J. S.; Blackmond, D. G.; Strieter,
E. R., and Buchwald, S. L., "Reevaluation of the mechanism of the amination of
aryl halides catalyzed by BINAP-ligated palladium complexes," J. Am. Chem.
Soc. 2006. 128(11): p. 3584-3591.
Hamze, Abdallah; Provot, Olivier; Brion, Jean-Daniel, and Alami, Mouâd,
"Xphos ligand and platinum catalysts: A versatile catalyst for the synthesis of
functionalized [beta]-(E)-vinylsilanes from terminal alkynes," Journal of
Organometallic Chemistry 2008. 693(16): p. 2789-2797.
Khan, S. A., Microfluidic Synthesis of Colloidal Nanomaterials. 2006,
Massachusetts Institute of Technology.
Mason, W. P., Physical Acoustics. 1982: Academic Press.
Coakley, W. T., "Ultrasonic separations in analytical biotechnology," Trends in
Biotechnology 1997. 15(12): p. 506-511.
Hawkes, J. J.; Barber, R. W.; Emerson, D. R., and Coakley, W. T., "Continuous
cell washing and mixing driven by an ultrasound standing wave within a
microfluidic channel," Lab Chip 2004. 4(5): p. 446-452.
Thompson, L. H. and Doraiswamy, L. K., "Sonochemistry: Science and
engineering," Industrial & Engineering Chemistry Research 1999. 38(4): p. 12151249.
Mason, T. J., "Ultrasound in synthetic organic chemistry," Chem. Soc. Rev. 1997.
26(6): p. 443-451.
Barge, A.; Tagliapietra, S.; Tei, L.; Cintas, P., and Cravotto, G., "Pd-catalyzed
Reactions Promoted by Ultrasound and/or Microwave Irradiation," Current
Organic Chemistry 2008. 12(18): p. 1588-1612.
191
234.
235.
236.
237.
Johansson, Linda; Johansson, Stefan; Nikolajeff, Fredrik, and Thorslund, Sara,
"Effective mixing of laminar flows at a density interface by an integrated
ultrasonic transducer," Lab Chip 2009. 9(2): p. 297-304.
Yang, Z.; Goto, H.; Matsumoto, M., and Maeda, R., "Active micromixer for
microfluidic systems using lead-zirconate-titanate(PZT)-generated ultrasonic
vibration," Electrophoresis 2000. 21(1): p. 116-119.
Kreutzer, Michiel T; Günther, Axel, and Jensen, K. F, "Sample dispersion for
segmented flow in microchannels with rectangular cross section," Analytical
Chemistry 2008. 80(5): p. 1558-1567.
McMullen, Jonathan P.; Zaborenko, Nikolay; Papageorge, Alexander T., and
Jensen, Klavs F. Automated Microfluidic System for Online Optimization of a
Chemical Reaction Network. in AIChE Annual Meeting. 2007. Salt Lake City,
UT.
192
Appendix A.
Fabrication Details
The fabrication processes for the devices described in this thesis are presented as run
sheet tables. The run sheets provide the facility, equipment used, and cleanliness code for
each process step. Unless otherwise specified, devices were fabricated in one of the
Microsystem Technology Laboratories (MTL) at MIT. MTL consists of 3 facilities:
Integrated Circuits Laboratory (ICL), Technology Research Laboratory (TRL), and
Exploratory Materials Laboratory (EML). The cleanliness code describes the
compatibility of the process step, where brown is CMOS compatible, green indicates
metal-free silicon processing, yellow is KOH etch or Pyrex® compatible, and red is for
gold and III-V metals general processing. The equipment used is listed by its abbreviated
name in the CORAL scheduling and management software for the Microsystems
Technology Laboratory at MIT. The commonly machines and their abbreviations are
presented in Table A.1.
Table A.1. CORAL Abbreviations
Abbreviation
A2-WetOxBond
coater
diesaw
EV1
EV501
LAM490B
Postbake
Prebake
STS2
Equipment
MRL Industries Model 718 System Atmospheric Oxidation Furnace
Photoresist-depositing spincoater
Disco Abrasive System Model DAD-2H/6T
Electronic Visions EV620 Mask Aligner
Electronic Visions EV620-501 Wafer Aligner/Bonder
Lam Research Model 490B Reactive Ion Etch
Blue M Model DDC-146C (120°C)
Blue M Model DDC-146C (95°C)
ICP Deep Trench Etching System (6” wafers)
193
Table A.2. “Goldilocks” KOH-etched reactors
Start with low-stress nitride-coated wafers. Coat both sides of the wafer. Pattern the
channels on the front side and the ports on the backside. LAM-etch the nitride to
General process
expose the silicon. Then, KOH etch the silicon wafer to create flow channels and
ports. Next, anodically bond the Pyrex wafer to seal the flow channels. Finally,
deposit copper around the flow ports and diesaw the devices.
6-inch 650-μm-thick double-side polished silicon wafer with LPCVD 5,000 A
Starting material:
deposited silicon nitride (performed in ICL)
6-inch 650-μm-thick Pyrex wafer
STEP FAC
MACHINE
ACTION
CODE
NOTES
1
PATTERN FLOW PORTS AND CHANNELS
1.1
TRL
HMDS
Coat wafer with HMDS
1.2
TRL
coater
Spincoat OCG 825 on the backside to define ports
1.3
TRL
prebake
Bake at 95°C for 15 minutes
1.4
TRL
coater
Spincoat OCG 825 on the front side to define channels
1.5
TRL
prebake
Bake at 95°C for 25 minutes
1.6
TRL
EV1
Expose backside for 1.5 seconds
1.7
TRL
photowet-1
Develop OCG 934 1:1, 7 seconds
1.8
TRL
EV1
Expose front side for 1.5 seconds
1.9
TRL
photowet-1
Develop OCG 934:1:1, 1-3 minutes
1.10
TRL
postbake
Postbake at 120°C for 30 minutes
1.11
ICL
LAM-490B
Etch exposed nitride
1.12
TRL
acidhood
piranha clean wafer
2
Program 4
square port mask
channel mask
“nitride on Si” recipe
ETCH FLOW PORTS AND CHANNELS
2.1
ICL
2.2
TRL
TMAH-KOH hood 25%, 80°C KOH etch wafer to ~400 um
~7 hr
post KOH clean and nitride removal (2 piranhas; 50:1
acidhood
HF dip for 30 min)
3
NITRIDE COATING
3.1
TRL
RCA
RCA clean wafer
3.2
ICL
tube-6D
grow CVD nitride ~0.5 um
4.1
TRL
acidhood
piranha Pyrex and silicon wafers
4.2
TRL
EV501-620
Bond Pyrex and silicon wafers
4
~ 3.5 hours
BOND PYREX WAFER
5
DEPOSIT COPPER
Only needed for
solder packaging
5.1
TRL
photoroom
Align shadow mask
5.2
TRL
ebeamAu
Deposit 100nm Ti and 500nm of Copper
6
ICL
diesaw
DIESAW DEVICES
194
(a)
(b)
(c)
Figure A.1. Goldilocks reactor masks: (a) channels; (b) bond pads; (c) ports.
The fabrication process for the KOH-etched meso-scale reactor is nearly identical to
that for the Goldilocks devices presented in Table A.2, with the following differences.
The starting silicon wafer is 1-mm thick. Step 2.1 is performed to a 750-μm depth, for
~12.5 hours. Step 5 (metal deposition) is not performed.
195
(a)
(b)
Figure A.2. KOH-etched meso-reactor masks: (a) channel; (b) ports.
Table A.3. HNA-etched mesoreactor
Start with low-stress nitride-coated wafers. Coat both sides of the wafer with thick
resist. Pattern the channels on the front side. LAM-etch the nitride to expose the
General process
silicon. Then, acid wet-etch the silicon wafer to create flow channels. In a Pyrex wafer,
drill inlet and outlet ports using an excitomer laser. Next, anodically bond the Pyrex
wafer to seal the flow channels.
6-inch 1000-μm-thick double-side polished silicon wafer with LPCVD 20,000 A
Starting material:
deposited silicon nitride (performed in ICL)
6-inch 1000-μm-thick Pyrex wafer
STEP FAC
MACHINE
ACTION
NOTES
CODE
1
1.1
PATTERN FLOW PORTS AND CHANNELS
TRL
HMDS
Coat wafer with HMDS
Program 3
10 μm thick
1.2
TRL
coater
Spincoat AZ 9260 on the front side to define channels
1.3
TRL
prebake
Bake at 95°C for 30 minutes
1.4
TRL
EV1
Expose front side 3×17 seconds with 15-second rest intervals channel mask
1.5
TRL
photowet-1
Develop in AZ 440
1.6
TRL
prebake
Bake at 95°C for 60 minutes
1.7
ICL
LAM-490B
Etch exposed nitride
2.1
TRL
acidhood
6:3:1 HF/HNO3/HAc mixture (600 μm)
~6 min, with stirring
2.2
TRL
acidhood
HF etch to strip nitride layer
~6 hours
2
“2μm nitride” recipe
ETCH FLOW CHANNELS
3
OXIDE COATING
3.1
TRL
RCA
3.2
TRL
A2-WetOxBond grow wet oxide ~0.5 um
4
4.1
~3 minutes
TRL
photoroom
RCA clean
~ 1.5 hours
DRILL PYREX WAFER
Optional
Spincoat AZ P 460 (thick resist)
Particle protection
196
4.2
TRL
prebake
Bake at 95°C for 30 minutes
4.3
EML Resonetics
5
Drill two holes for in/out ports
BOND PYREX WAFER
5.1
TRL
acidhood
piranha Pyrex and silicon wafers
5.2
TRL
EV501-620
Bond Pyrex and silicon wafers
Figure A.3. HNA-etched meso-reactor mask.
Table A.4. First-generation micromixer
General
process:
The wafers are oxidized to act as a mask for shallow KOH etching of the backside flow
channels. The front side is then patterned and etched to reveal the major flow channels
through the oxide layer. Photoresist protects the exposed Si to pattern through holes for
connecting to the backside flow channels. DRIE is used to partially etch these holes, the
photoresist is then removed, and the etching process finishes the through holes and
defines the main flow channels. Lastly, two anodic bonds using Pyrex wafers are used to
seal the flow channels.
Materials:
6-inch 650-μm-thick double-side polished silicon wafer
6-inch Pyrex predrilled wafer (Bullen Ultrasonics)
6-inch 650-μm-thick Pyrex wafer
STEP
FAC
MACHINE
1
ACTION
NOTES
OXIDIZE WAFER
1.1.1
TRL RCA
RCA clean wafer
1.1.2
TRL A2-WetOxBond
Grow 5000 Å wet oxide
197
90 minutes
CODE
2
DEFINE BACKSIDE
CHANNELS
2.1
Photolithography
2.1.1
TRL HMDS
Coat wafer with HMDS
2.1.2
TRL coater
Spincoat OCG 825 to define
backside channels
2.1.3
TRL prebake
Bake at 95°C for 30 minutes
2.1.4
TRL EV1
Expose resist for 1.5 seconds (dP
Mask 1
channel mask)
2.1.5
TRL photowet-1
Develop OCG 934 1:1
2.1.6
TRL postbake
Postbake at 120°C for 30 minutes
2.1.7
TRL acidhood2
BOE patterned SiO2
2.1.8
TRL acidhood2
piranha clean wafer
2.2
1-3 min
Need ~ 5 min in BOE
KOH Etch channels
2.2.1
ICL TMAH-KOH hood
25%, 70°C KOH etch wafer to
~35 µm
2.2.2
TRL acidhood
post KOH clean (2 piranhas; 50:1
HF dip)
3.1.1
TRL RCA
RCA clean wafer
3.1.2
TRL A2-WetOxBond
Grow 10,000 Å wet oxide
3
~1 hr
OXIDIZE WAFER
200 minutes
DEFINE FRONTSIDE
CHANNELS & THRUHOLES
4
4.1
Define front side channels
4.1.1
TRL HMDS
Coat wafer with HMDS
4.1.2
TRL coater
Spincoat OCG 825 to define
front-side channels
4.1.3
TRL prebake
Bake at 95°C for 30 minutes
4.1.4
TRL EV1
Expose resist for 1.5 seconds
(main channel mask)
Mask 2
4.1.5
TRL photowet-1
Develop OCG 934 1:1
1-3 min
4.1.6
TRL postbake
Postbake at 120°C for 30 minutes
4.1.7
TRL acidhood2
BOE patterned SiO2
4.1.8
TRL acidhood2
piranha clean wafer
4.2
Define through holes
4.2.1
TRL HMDS
Coat wafer with HMDS
4.2.2
TRL coater
Double spincoat thick photoresist
~20 mm
AZP4620 on front side
198
4.2.3
TRL prebake
Bake at 95°C for 30+ minutes
4.2.4
TRL EV1
Expose resist for 40 seconds to
UV (Flowthrough Mask)
Mask 3
4.2.5
TRL photowet-1
Development (AZ 440)
5 min
4.2.6
TRL prebake
Postbake at 95°C for 30 minutes
4.3
Attach handle wafer
4.3.1
TRL coater
spincoat thick resist on handle
wafer, attach wafer 1
4.3.2
TRL prebake
Postbake at 95°C 30 min
4.4
use "target" pattern to attach
wafers
STS etch through holes
4.4.1
TRL STS2
STS etch, recipe ole3, 650 um
4.4.2
TRL acidhood
piranha clean wafer
4.5
~7 hours
Attach handle wafer
4.5.1
TRL coater
spincoat thick resist on handle
wafer, attach wafer 1
4.5.2
TRL prebake
Postbake at 95°C 30 min
"target" pattern
STS etch through holes and main
channels
4.6
4.6.1
TRL STS2
STS etch, recipe ole3, 200 µm
4.6.2
TRL acidhood
piranha clean wafer
4.6.3
TRL acidhood2
BOE etch to remove oxide
4.7
~2.2 hours
Oxidize wafer
4.7.1
TRL RCA
RCA clean wafer
4.7.2
TRL tube-a2
Grow 2000 Å wet oxide
5
DEVICE PACKAGING
5.1
CAP DEVICE
5.1.1
TRL acidhood
Piranha clean wafers
5.1.2
TRL EV620
Align and bond predrilled-Pyrex
and Si wafers
5.1.3
TRL EV620
Align and bond wafer stack and
Pyrex bottom wafer
5.2
20 minutes
Touch flags to Si wafer and move
aside the flag removers
Dice device
5.2.1
ICL diesaw
score devices along wide section cut 700-μm deep
5.2.2
ICL diesaw
dice devices
cut 700-μm deep
199
(a)
(b)
(c)
Figure A.4. First-generation micromixer masks: (a) channels; (b) manifolds/mixer; (c) throughholes.
Table A.5. Second-generation micromixer
General process
Start with fresh wafers. Coat both sides of the wafer with thick resist. Pattern the
channels/mixer cavity on the front side, and ports/through-holes on the backside. DRIE
the front side to 200 μm. Mount the wafer on a handle wafer and DRIE the throughholes. Next, anodically bond the Pyrex wafer to seal the flow channels.
6-inch 650-μm-thick double-side polished silicon
6-inch 650-μm-thick Pyrex wafer
MACHINE
ACTION
Starting material:
STEP FAC
1
NOTES
PATTERN FLOW PORTS AND CHANNELS
1.1
TRL
HMDS
Coat wafer with HMDS
Program 3
1.2
TRL
coater
Spincoat AZ 9260 on the front side to define channels
10 μm thick
1.3
TRL
prebake
Bake at 95°C for 15 minutes
1.4
TRL
coater
Spincoat AZ 9260 on the backside
1.5
TRL
prebake
Bake at 95°C for 15 minutes
1.6
TRL
coater
Spincoat AZ 9260 on the backside
1.7
TRL
prebake
Bake at 95°C for 30 minutes
1.8
TRL
EV1
Expose front side 3×17 seconds with 15-second rest intervals channel/mixer mask
1.9
TRL
photowet-1
Develop in AZ 440
1.10
TRL
EV1
Expose back side 7×17 seconds with 15-second rest intervals through hole mask
1.11
TRL
photowet-1
Develop in AZ 440
1.12
TRL
prebake
Bake at 95°C for 60 minutes
10 μm thick
additional 10 μm
~15 seconds
~5 minutes
200
CODE
2
DRIE ETCH (STS)
2.1
STS etch main flow channels
2.1.1
TRL
STS2
STS etch, recipe Kishori, 200 μm
2.2
~130 min.
2.2.1
TRL
coater
Attach handle wafer
spincoat thick resist on handle wafer, attach Si wafer by front use "target" pattern
side
to attach wafers
2.2.2
TRL
prebake
Postbake at 95°C 30 min
2.3
STS etch through holes
2.3.1
TRL
STS2
STS etch, recipe Kishori, 450 μm (until all the way through) ~5 hours
2.3.2
TRL
acidhood
piranha clean wafer
3.1
TRL
RCA
3.2
TRL
A2-WetOxBond grow wet oxide ~0.5 μm
3
OXIDE COATING
4
RCA clean
~ 1.5 hours
BOND PYREX WAFER
4.1
TRL
acidhood
piranha Pyrex and silicon wafers
4.2
TRL
EV501-620
Bond Pyrex and silicon wafers
(a)
(b)
Figure A.5. Second-generation micromixer masks: (a) channels/mixer; (b) ports/through-holes.
The fabrication processes for the single-addition spiral microreactors discussed in
section 6.4.2 (Figures 6.5 and 6.7) is nearly identical to that for the second-generation
micromixer devices presented in Table A.5, with the following differences. For the 120μL device of Figure 6.5, the front side is coated with two layers of resist, while the
backside is coated with one layer (step 1.4 applies to the front side; thus, exposure times
in steps 1.8 and 1.10 are switched); in steps 2.1.1 and 2.3.1 (the front and back etches),
the etch depths are 400 μm and 250 μm, respectively. For the 220-μL device of Figure
201
6.7, the starting silicon and Pyrex wafers are 1-mm thick. Both the front and backsides
are coated with two layers of resist, exposed as in step 1.10, and etched to a depth of 500
μm. In Figure A.6, the halo etch (oblong closed channel) shown in part (c) is present in
the corresponding places on the front-side masks (a) and (b), but was not shown due to
space constraints.
(a)
(b)
(c)
Figure A.6. Spiral single-addition reactors: (a) 120-μL, 400×400 μm channel device; (b) 220-μL,
500×500 μm channel device; (c) ports and halo through etch.
Table A.6. Spiral sequential-addition reactor
Oxidize the wafers to act as a mask for shallow DRIE etching of the sequential addition
channels. Pattern the front side and etch to reveal the sequential addition channels
through the oxide layer. Pattern the front side and etch to reveal the main flow channel,
General process: which is then etched by DRIE. Remove the photoresist is then removed and etch the
sequential addition channels. Mount the wafer and etch by DRIE the backside through
holes. Deposit nitride to act as a protective layer. Lastly, bond to a Pyrex wafers to seal the
flow channels.
Materials:
6-inch 650-μm-thick double-side polished silicon wafer
6-inch 650-μm-thick Pyrex wafer
202
STEP
FAC
MACHINE
1
ACTION
NOTES
OXIDIZE WAFER
1.1
TRL
RCA
RCA clean wafer
1.2
TRL
A2-WetOxBond
Grow 2000 Å wet oxide
30 minutes
DEFINE SEQUENTIAL
ADDITION CHANNELS
2
2.1
TRL
HMDS
Coat wafer with HMDS
2.2
TRL
coater
Spincoat OCG 825 to define
sequential addition channels
2.3
TRL
prebake
Bake at 95°C for 30 minutes
2.4
TRL
EV1
Expose resist for 1.5 seconds
sequential channels mask
2.5
TRL
photowet-1
Develop in OCG 934 1:1
1-3 min
2.6
TRL
postbake
Postbake at 120°C for 30 minutes
2.7
TRL
acidhood2
BOE patterned SiO2
2.8
TRL
acidhood2
piranha clean wafer
~2 min in BOE
DEFINE MAIN CHANNELS AND
THROUGH-HOLES
3
3.1
1 μm
TRL
coater
Coat wafer with HMDS
Program 3
Spincoat AZ 9260 on the front side
to define channels
10 μm thick
3.3
TRL
prebake
Bake at 95°C for 15 minutes
3.4
TRL
coater
Spincoat AZ 9260 on the front side additional 10 μm
3.5
TRL
prebake
Bake at 95°C for 15 minutes
3.6
TRL
coater
Spincoat AZ 9260 on the backside
3.7
TRL
prebake
TRL
EV1
Bake at 95°C for 30 minutes
Expose front side 7×17 seconds with
15-second rest intervals
main channel mask
TRL
photowet-1
TRL
EV1
TRL
TRL
photowet-1
acidhood2
3.2
3.8
3.9
3.10
3.11
3.12
TRL
HMDS
4
Develop in AZ 440
~15 seconds
Expose back side 3×17 seconds with
15-second rest intervals
through hole mask
Develop in AZ 440
BOE patterned SiO2
~5 minutes
~2 min in BOE
ETCH WAFER
4.1
4.1.1
10 μm thick
STS etch through holes
TRL
STS2
4.2
4.2.1
TRL
coater
4.2.2
TRL
prebake
STS etch, recipe Kishori, 300 μm
~4 hours
Attach handle wafer
spincoat thick resist on handle wafer, use "target" pattern to attach
attach wafer 1 by backside
wafers
Postbake at 95°C 30 min
203
CODE
4.3
STS etch main channel
4.3.1
TRL
STS2
4.3.2
TRL
acidhood
4.4
4.4.1
TRL
coater
4.4.2
TRL
prebake
STS etch, recipe Kishori, 390 μm
piranha clean wafer
~4.5 hours
Attach handle wafer
spincoat thick resist on handle wafer, use "target" pattern to attach
attach wafer 1 by backside
wafers
Postbake at 95°C 30 min
STS etch main and sequential
addition channels
4.5
4.5.1
TRL
STS2
4.5.2
TRL
acidhood
STS etch, recipe Kishori, 10.5 μm
piranha clean wafer
4.5.3
TRL
acidhood2
1:10 HF dip to remove oxide
5
~7 minutes
NITRIDE COATING
5.1
TRL
RCA
RCA clean wafer
5.2
ICL
tube-6D
grow CVD nitride ~0.5 um
6.1
TRL
acidhood
piranha Pyrex and silicon wafers
6.2
TRL
EV501-620
Bond Pyrex and silicon wafers
7
ICL
diesaw
DIESAW DEVICES
6
~ 3.5 hours
BOND PYREX WAFER
(a)
(b)
Figure A.7. Spiral sequential-addition reactor: (a) main channel mask; (b) sequential addition
channel mask.
The sequential addition reactor mask for the through-etch ports and halo is identical
to the mask shown in Figure A.6c.
204